´
F. Boratynski et al. / Food Chemistry 141 (2013) 419–427
422
OH of 2a); 3.88 (ddd, J = 9.0, 5.5, 4.8 Hz, 1H, H-5 of 2a); 4.12 (dt,
J = 8.3, 5.3 Hz, 1H, H-5 of 3a); 5.43 (dd, J = 5.6, 2.6 Hz, 1H, H-2 of
2a); 5.52 (dd, J = 5.4, 2.9 Hz, 1H, H-2 of 3a); 13C NMR (150 MHz,
CDCl3): d: 14.06 (CH3-4 of 3a), 14.51 (C-40 of 3a), 14.52 (C-40 of
2a), 14.75 (CH3-4 of 2a), 22.70, 22.84, 28.72, 29.71, 30.04, 30.89
(C-10, C-20, C-30 both isomers), 34.41 (C-4 of 3a), 34.81 (C-4 of
2a), 41.50 (C-3 of 2a), 42.12 (C-3 of 3a), 80.72 (C-5 of 3a), 82.97
(C-5 of 2a), 97.41 (C-2 of 3a), 98.46 (C-2 of 2a); IR (film, cmꢀ1):
3408 (m), 1456 (m), 1062 (m).
CH2-10, CH2-20, CH2-30 both isomers); 1.80 (m, 1H, H-4 of 3b);
2.06 (dd, J = 12.5, 6.7 Hz, 1H, one of CH2-3 of 2b); 2.13 (m, 1H, H-
4 of 2b); 2.38 (ddd, J = 14.0, 9.0, 5.6 Hz, 1H, one of CH2-3 of 3b);
2.59 (s, 1H, OH of 2b); 2.75 (s, 1H, OH of 3b); 3.48 (dt, J = 8.3,
4.0 Hz, 1H, H-5 of 2b); 3.69 (dt, J = 8.0, 3.6 Hz, 1H, H-5 of 3b);
5.42 (m, 1H, H-2 of 2b); 5.50 (m, 1H, H-2 of 3b); IR (film, cmꢀ1):
3408 (m), 1456 (m), 1062 (m).
2.7. Extraction procedure using a Deryng apparatus
2.6.2.4. Preparative oxidation of ( )-1b by PADH II. Oxidation of ( )-
1b (0.1 g), after 48 h, gave a mixture of: unreacted ( )-diol 1b
(41%), (ꢀ)-(4R,5S)-4b (56%, ee = 82%) and mixture of hemiacetals
2b and 3b (3%). The column chromatography of this mixture affor-
After the biotransformation, the combined aqueous fractions
were placed in a 500 ml round flask and 0.1 M NaOH was added
portionwise to pH = 12. Sample flask was heated for 2 h, after the
boiling point was reached. The vapours were condensed by means
of a cold refrigerant. The main purpose of medium alkalization was
conversion of obtained lactones 4a and 4b, by opening of lactone
ring, into hydroxyacid salts, which are water-soluble and non-vol-
atile compounds. After the extraction, the solvent, 1 ml of cyclo-
hexane, containing the volatile compounds – hemiacetals 2a–3a
or 2b–3b, was collected in a 2.5 ml vial. Then, the reaction mixture
was acidified by 0.1 M HCl to pH = 3 and distilled again for 2 h in
Deryng apparatus. During the hydrodistillation of volatile com-
pounds, contained in the aqueous layer, whisky lactones 4a and
4b were extracted with 1 ml of cyclohexane. The unreacted diol
1a and 1b, remained in the aqueous medium, was used to next
enzymatic oxidation. In this way, volatile products were separated
from substrate 1a without time-consuming and toxic solvent-free
column chromatography.
ded 0.044 g (44% yield) of (ꢀ)-(4R,5S)-4b, ee = 82%, ½a D20
ꢂ
¼ ꢀ84ꢃ (c
0.2, CHCl3), ((Wilkinson et al., 2004): (ꢀ)-(4R,5S)-4b ee > 99%,
½
a 2D0
ꢂ
¼ ꢀ97ꢃ (c 0.34, CH3OH)).
2.6.2.5. Preparative oxidation of ( )-1b by HLADH. Oxidation of ( )-
1b (0.1 g), after 24 h, gave a mixture of: unreacted ( )-diol 1b
(55%), (ꢀ)-(4R,5S)-4b (31%, ee = 27%) and mixture of hemiacetals
2b and 3b (14%). The column chromatography of this mixture
afforded 0.028 g (28% yield) of (ꢀ)-(4R,5S)-4b, ee = 27%,
½
a 2D0
ꢂ
¼ ꢀ8:4ꢃ (c 5.2, CHCl3) and 0.012 g (12%) of hemiacetals 2b
and 3b. The oxidation of ( )-1b for longer time (120 h) let to obtain
( )-4b as the only product in high (87%) yield.
2.6.2.6. Preparative oxidation of ( )-1b by HLADH recombinant from
E. coli. Oxidation of ( )-1b (0.1 g), after 24 h, gave a mixture of:
unreacted ( )-diol 1b (24%), (ꢀ)-(4R,5S)-4b (60%, ee = 34%) and
mixture of hemiacetals 2b and 3b (16%). The column chromatogra-
phy of this mixture afforded 0.051 g (51% yield) of (ꢀ)-(4R,5S)-4b,
2.8. Oxidation of erythro-3-methylnonane-1,4-diol (1a) with TEMPO/
BAIB
ee = 34%, ½a 2D0
ꢂ
¼ ꢀ8:7ꢃ (c 6.8, CHCl3), 0.015 g (15%) of hemiacetals
2b and 3b and 0.02 g (20% yield) of unreacted ( )-diol 1b. The oxi-
dation of ( )-1b for longer time (120 h) let to obtain ( )-4b as the
only product in high (92%) yield.
To a stirred room temperature solution of diol 1a (0.025 g,
0.04 mmol), dissolved in methylene chloride (3 ml) was added
sequentially bis-acetoxyiodobenzene (BAIB, 0.014 g, 0.04 mmol)
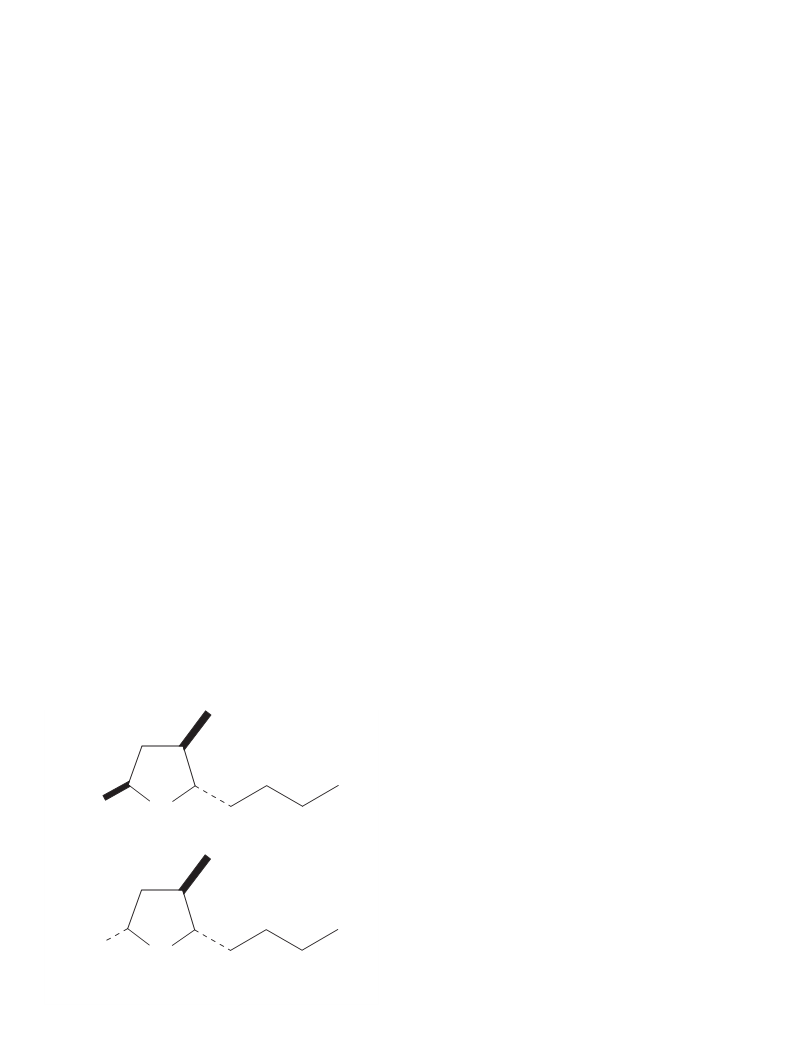
2.6.2.6.1. Trans-5-butyl-4-methyltetrahydrofuran-2-ols (mixture of
isomers; 2b and 3b) (Fig. 2). Colorless liquid, trans (3b) and cis
(2b) isomers of the composition 53 : 47%; 1H NMR (600 MHz,
CDCl3) d: 0.90 (t, J = 6.9 Hz, 3H, CH3-40 of 2b); 0.91 (t, J = 6.9 Hz,
3H, CH3-40 of 3b); 1.03 (d, J = 6.5 Hz, 3H, CH3-4 of 2b); 1.06 (d,
J = 6.7 Hz, 3H, CH3-4 of 3b); 1.23–1.68 (m, 14H, one of CH2-3,
and
2,2,6,6-tetramethylpiperidinooxy
(TEMPO,
0.001 g,
0.003 mmol). The reaction was conducted at room temperature.
Progress of the reaction was monitored by TLC and GC. After 4 h
of the reaction, a saturated solution of Na2S2O3 (10 ml) was added
and the mixture was extracted with diethyl ether (25 ml). The or-
ganic fraction was separated and washed with saturated NaHCO3
(10 ml) solution and then, water (10 ml). The combined aqueous
layers were extracted three times with diethyl ether (25 ml). Ether
extract was washed with brine, dried over anhydrous MgSO4, fil-
tered and concentrated by rotary evaporation. After GC analysis
with application of chiral column racemic mixture of lactone 4a
was obtained.
3
4
2'
4'
2
5
1
HO
HO
O
1'
3'
3b
2.9. Oxidation of mixture of isomers cis-5-butyl-4-
methyltetrahydrofuran-2-ols (2a and 3a) with pyridine dichromate
In a round bottom flask a mixture of diastereomeric hemiacetals
2a and 3a (0.05 g, 0.4 mmol), pyridine dichromate (0.3 g,
0.8 mmol) and anhydrous sodium acetate (0.022 g, 0.2 mmol) dis-
solved in anhydrous methylene chloride (20 ml) were placed. The
reaction was carried out at room temperature with continuous
stirring. Progress of the reaction was monitored by TLC and GC
analysis. After 4 h of the reaction, solvent was evaporated, and
the residue was extracted with hexane. The organic layer was fil-
tered through Florisil, dried over anhydrous MgSO4 and evaporated
in vacuo to afforded 0.04 g (80% yield) of (ꢀ)-(4S,5S)-4a, ee = 38%,
3
4
2'
4'
2
5
1
O
2b
1'
3'
Fig. 2. Trans-5-butyl-4-methyltetrahydrofuran-2-ols (mixture of isomers; 2b and
3b).
½
a 2D0
ꢂ
¼ ꢀ23:1ꢃ (c 5.2, CHCl3).

 Boratyński, Filip
Boratyński, Filip