Y.-Z. Xu, Q.-L. Wen, F. Sha et al.
Tetrahedron 92 (2021) 132273
was dissolved in DMF (41 mL) and MeOH (12 mL) before DMAP
EtOAc, and washed with water. The mixture was extracted with
EtOAc (20 mL x 3). The combined organic layers were washed with
brine (10 mL) and dried over Na SO , filtered and concentrated
2 4
under reduced pressure. Purification of the residue by flash column
chromatography (20/1 PE/EtOAc) afforded compound 19 (372.6 mg,
(
994.5 mg, 8.14 mmol) was added. The mixture was heated for 4 h
ꢀ
at 50 C. Upon cooling, water and EtOAc were added, and the
organic layer was separated. The aqueous layer was extracted with
EtOAc (60 mL x 3). The combined organic layers were washed with
brine and dried over Na
2
SO
4
, filtered and concentrated under
80%) as a pale yellow syrup.
1
reduced pressure afforded the crude product, which was further
purified by flash chromatography (50/1 PE/EtOAc) afforded com-
pound 16 (7.41 g, 75%, 3 steps) as a pale yellow solid.
H NMR (400 MHz, CDCl
3
)
d
7.76 (d, J ¼ 2.4 Hz, 1H), 7.55 (dd,
J ¼ 8.4 Hz, 2.4 Hz, 1H), 7.50 (d, J ¼ 8.8 Hz, 2H), 7.39e7.29 (m, 5H),
7.06 (d, J ¼ 8.8 Hz, 1H), 6.95 (d, J ¼ 8.8 Hz, 2H), 6.43 (d, J ¼ 10.0 Hz,
1H), 6.26 (d, J ¼ 2.4 Hz, 1H), 6.18 (d, J ¼ 2.4 Hz, 1H), 5.43 (d,
J ¼ 10.0 Hz, 1H), 4.95 (s, 2H), 3.84 (s, 3H), 3.58 (s, 3H), 3.23 (s, 3H),
1.39 (s, 6H).
ꢀ
m.p. 67.8e68.8 C
1
H NMR (400 MHz, CDCl
3
)
d
7.98 (d, J ¼ 2.4 Hz,1H), 7.61 (brs,1H),
7
2
.24 (dd, J ¼ 8.8 Hz, 2.4 Hz, 1H), 6.55 (d, J ¼ 8.8 Hz, 1H), 3.85 (s, 3H),
.88 (s, 3H).
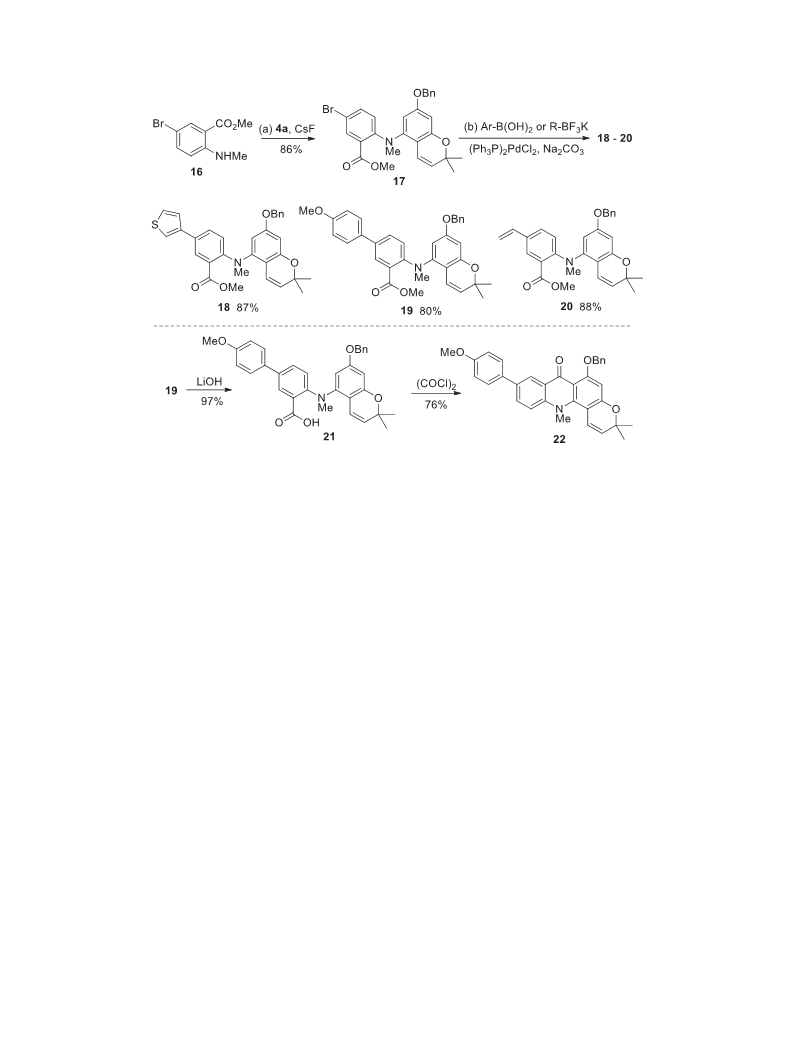
.2.18 Gram-scale synthesis of compound 17To a stirred solution
of silyltriflate 4a (4.87 g, 10.00 mmol) and compound 16 (2.56 g,
0.50 mmol) in DME (100 mL) was added CsF (4.56 g, 30.00 mmol).
13C NMR (151 MHz, CDCl
3
) d 168.7, 159.9, 158.9, 155.4, 148.0,
4
147.7,136.8,133.2,132.4,129.9,129.1,128.5,127.9,127.6,127.0,124.3,
127.0, 124.3, 120.7, 119.4, 116.0, 114.7, 114.2, 110.2, 103.1, 98.6, 75.6,
70.0, 55.3, 51.8, 42.5, 27.4.
1
ꢀ
The solution was stirred at 80 C for 24 h, then cooled to room
temperature, filtered over silica gel (CH Cl eluent). Evaporation
under reduced pressure afforded the crude product, which was
IR (ATR): nmax 2957, 1697, 1605, 1558, 1486, 1435, 1343, 1240,
ꢁ
1135, 1111, 1034, 825, 739, 696 cm
1
2
2
HRMS-ESI (m/z): [MþH]þ calcd for C34
H34NO 536.2431, found
þ
5
further purified by flash chromatography (100/1 / 80/1 PE/EtOAc)
536.2436.
afforded compound 17 (4.40 g, 86%) as a pale yellow syrup.
4.2.21 Synthesis of compound 20 To a Schlenk flask were added
compound 17 (325.0 mg, 0.639 mmol), potassium vinyl-
1
H NMR (400 MHz, CDCl
3
)
d
7.64 (d, J ¼ 2.4 Hz, 1H), 7.42 (dd,
J ¼ 8.8 Hz, 2.4 Hz, 1H), 7.38e7.29 (m, 5H), 6.87 (d, J ¼ 8.8 Hz, 1H),
trifluoroborate (256.8 mg, 1.917 mmol) and (Ph
0.032 mmol). The flask was degassed with N
Na CO (2 M aqueous, 1.28 mL, 2.56 mmol) and dioxane (15 mL)
3
2 2
P) PdCl (22.5 mg,
6
1
1
.37 (d, J ¼ 9.6 Hz, 1H), 6.32 (d, J ¼ 2.4 Hz, 1H), 6.12 (d, J ¼ 2.4 Hz,
H), 5.45 (d, J ¼ 9.6 Hz, 1H), 4.93 (s, 2H), 3.54 (s, 3H), 3.16 (s, 3H),
2
for 3 times, then
2
3
ꢀ
.39 (s, 6H).
were added to the flask, and then stirred for 16 h at 100 C. The
reaction mixture was cooled to room temperature, diluted with
EtOAc, and washed with water. The mixture was extracted with
EtOAc (15 mL x 3). The combined organic layers were washed with
13C NMR (101 MHz, CDCl
36.7, 134.6, 133.5, 128.5, 127.9, 127.5, 127.4, 125.3, 121.6, 119.1, 112.6,
10.3, 103.3, 91.1, 75.7, 70.0, 51.9, 42.2, 27.5.
IR (ATR): max 2972, 1718, 1603, 1477, 1432, 1388, 1288, 1243,
3
) d 167.1, 159.9, 155.5, 148.4, 147.1,
1
1
n
2 4
brine (10 mL) and dried over Na SO , filtered and concentrated
under reduced pressure. Purification of the residue by flash column
chromatography (30/1 PE/EtOAc) afforded compound 20
ꢁ
1
1
133, 1111, 1027, 812, 732, 695 cm
HRMS-ESI (m/z): [MþH]þ calcd for C27
H27BrNO
þ
4
510.1098,
found 510.1102.
(255.5 mg, 88%) as a pale yellow syrup.
1
4
.2.19 Synthesis of compound 18 To a Schlenk flask were added
H NMR (400 MHz, CDCl
3
)
d
7.59 (d, J ¼ 2.4 Hz, 1H), 7.42e7.30
compound 17 (325.0 mg, 0.639 mmol), 3-thiopheneboronic acid
(m, 6H), 6.96 (d, J ¼ 8.8 Hz, 1H), 6.64 (dd, J ¼ 17.6 Hz, 10.8 Hz, 1H),
6.39 (d, J ¼ 10.0 Hz, 1H), 6.26 (d, J ¼ 2.4 Hz, 1H), 6.14 (d, J ¼ 2.4 Hz,
1H), 5.65 (d, J ¼ 17.6 Hz, 1H), 5.43 (d, J ¼ 10.0 Hz, 1H), 5.17 (d,
(
245.3 mg, 1.917 mmol) and (Ph
3
P)
2
PdCl
2
(22.5 mg, 0.032 mmol).
CO (2 M
The flask was degassed with N
2
for 3 times, then Na
2
3
aqueous, 1.28 mL, 2.56 mmol) and dioxane (15 mL) were added to
J ¼ 10.8 Hz, 1H), 4.94 (s, 2H), 3.55 (s, 3H), 3.20 (s, 3H), 1.39 (s, 6H).
ꢀ
13
the flask, and then stirred for 18 h at 100 C. The reaction mixture
3
C NMR (151 MHz, CDCl ) d 168.6, 159.9, 155.4, 148.7, 147.5,
was cooled to room temperature, diluted with EtOAc, and washed
with water. The mixture was extracted with EtOAc (15 mL x 3). The
combined organic layers were washed with brine (10 mL) and dried
136.7,135.5,130.1,129.5,129.0,128.5,127.9,127.5,127.1,123.8,120.0,
119.3, 112.5, 110.3, 103.2, 98.8, 75.6, 70.0, 51.8, 42.5, 27.4.
IR (ATR): nmax 2964, 1715, 1604, 1493, 1434, 1350, 1295, 1243,
ꢁ
1
over Na
2
SO
4
, filtered and concentrated under reduced pressure.
1197, 1133, 1114, 1030, 827, 696 cm
HRMS-ESI (m/z): [MþH]þ calcd for C29
H30NO 456.2169, found
þ
4
Purification of the residue by flash column chromatography (30/1
PE/EtOAc) afforded compound 18 (286.1 mg, 87%) as a pale yellow
syrup.
456.2176.
4.2.22 Synthesis of compound 21 To a solution of compound 19
(425.8 mg, 0.80 mmol) in MeOH/THF/H O (8.8 mL, 5/5/1) was
added LiOH∙H O (335.7 mg, 8.0 mmol). The resulting mixture was
1H NMR (400 MHz, CDCl
)
d
7.80 (d, J ¼ 2.0 Hz, 1H), 7.58 (dd,
3
2
J ¼ 8.8 Hz, 2.0 Hz, 1H), 7.39e7.28 (m, 8H), 7.03 (d, J ¼ 8.8 Hz, 1H),
2
6
1
1
.42 (d, J ¼ 10.0 Hz, 1H), 6.27 (d, J ¼ 2.0 Hz, 1H), 6.17 (d, J ¼ 2.0 Hz,
H), 5.43 (d, J ¼ 10.0 Hz, 1H), 4.94 (s, 2H), 3.57 (s, 3H), 3.22 (s, 3H),
heated under reflux for 12 h. The reaction mixture was subse-
quently allowed to cool at room temperature and acidified with 2 N
HCl (aq). After separating both layers, the aqueous layer was
extracted with EtOAc (5 mL x 3). The combined organic layers were
.39 (s, 6H).
13C NMR (151 MHz, CDCl
41.0, 136.7, 129.7, 128.9, 128.5, 128.4, 127.9, 127.5, 127.0, 126.2,
25.9,124.1,120.5,119.4, 110.2,103.1, 98.7, 75.6, 69.9, 51.8, 42.4, 27.4.
IR (ATR): max 2943, 1713, 1603, 1565, 1486, 1434, 1296, 1236,
3
) d 168.5, 159.9, 155.4, 148.2, 147.6,
1
1
2 4
dried with Na SO and the solvent was evaporated under reduced
pressure. Purification of the residue by flash column chromatog-
raphy (4/1 PE/EtOAc) provided compound 21 (402.6 mg, 97%) as a
yellow solid.
n
ꢁ
134, 1112, 1029, 777, 695 cm
1
1
HRMS-ESI (m/z): [MþH]þ calcd for C31
H30NO
S
þ
512.1890,
m.p.75.9e77.0 C
ꢀ
4
1
found 512.1897.
H NMR (400 MHz, CDCl
3
)
d
13.98 (brs, 1H), 8.45 (d, J ¼ 1.6 Hz,
4
.2.20 Synthesis of compound 19 To a Schlenk flask were added
compound 17 (440.2 mg, 0.866 mmol), 4-methoxyphenylboronic
acid (394.8 mg, 2.598 mmol) and (Ph P) PdCl (30.4 mg,
.043 mmol). The flask was degassed with N for 3 times, then
1H), 7.63 (dd, J ¼ 8.4 Hz, 1.6 Hz, 1H), 7.55 (d, J ¼ 8.4 Hz, 2H),
7.45e7.38 (m, 4H), 7.36e7.32 (m, 1H), 7.02 (d, J ¼ 8.4 Hz, 1H), 6.97
(d, J ¼ 8.8 Hz, 2H), 6.51 (d, J ¼ 2.4 Hz, 1H), 6.38 (d, J ¼ 2.4 Hz, 1H),
6.13 (d, J ¼ 10.0 Hz, 1H), 5.34 (d, J ¼ 10.0 Hz, 1H), 5.04 (s, 2H), 3.84 (s,
3
2
2
0
2
Na
2
CO
3
(2 M aqueous, 1.73 mL, 3.46 mmol) and dioxane (20 mL)
3H), 3.15 (s, 3H), 1.31 (s, 6H).
ꢀ
13
were added to the flask, and then stirred for 16 h at 100 C. The
3
C NMR (151 MHz, CDCl ) d 166.6, 159.6, 159.6, 155.7, 150.3,
reaction mixture was cooled to room temperature, diluted with
145.1, 138.7, 136.4, 132.0, 131.3, 130.5, 128.6, 128.3, 128.2, 128.0,
9

 Xu, Yuan-Ze
Xu, Yuan-Ze