132
Q.-X. Liu et al. / Journal of Molecular Structure 697 (2004) 131–135
(3.036 g, 0.13 mol) in 1, 4-dioxane (150 ml) and stirred for
1 h at 90 8C. Then 1, 4-dioxane (100 ml) solution of octane
bromide (22.210 g, 0.12 mol) was dropwise added to the
solution. The mixture was continued to stir for 48 h at 90 8C
and a yellow solution was obtained. The solvent was
removed with a rotary evaporator and H2O (500 ml) was
added to the residue. Then the solution was extracted with
CH2Cl2 (3 £ 100 ml), the extracting solution was dried
with anhydrous MgSO4. After removing CH2Cl2, a yellow
liquid 9-octylimidazole was obtained by distillation. Yield:
6.413 g (80%). bp: 125-127 8C. 1H NMR (300 MHZ,
CDCl3): 0.88 (t, J ¼ 6.6, 3H, CH3), 1.27 (m, 10H,
5 £ CH2), 1.78 (m, 2H, CH2), 3.94 (t, J ¼ 7.2, 2H, CH2),
6.92 (s, 1H, 4-imiH) (imi: imidazole), 7.06 (s, 1H, 5-imiH),
7.55 (s, 1H, 2-imiH).
(m, 2 £ 10H, imiH,PhH orAnH), 8.06 (d, J ¼ 8.0, 2 £ 2H,
AnH), 8.56 (d, J ¼ 8.0, 2 £ 2H, AnH), 8.79 (s, 2 £ 1H,
AnH). 13C NMR (75 MHz, CDCl3): 14.1 (CH3), 22.5 (CH2),
22.7 (CH2), 27.0 (CH2), 29.4 (CH2), 31.0 (CH2), 31.9 (CH2),
47.3 (NCH2CH2), 51.2 (NCH2An), 79.6 (CxCPh), 107.0
(PtCxC), 118.7 and 119.6, 120.5, 122.2, 122.8, 123.3,
124.3, 124.8, 125.3, 127.2, 128.3, 128.9, 131.3, 138.2 (AnC,
PhC or 4, 5-imiC), 172.0 (Ccarbene). IR (KBr; disk): n (CxC)
2096 (m) cm21
.
2.4. [1-(9-Anthracylmethyl)-3-ethylimidazol-2-
ylidene]2Pt(CxCPh)2 (2b)
This complex was prepared in a manner analogous to that
for 2a, only with 1-(9-anthracenylmethyl)-3-ethylimidazo-
lium iodide (1b) instead of 1-(9-anthracenylmethyl)-3-
octylimidazolium chloride (1a). The pale yellow powder
of [1-(9-anthracylmethyl)-3-ethylimidazol-2-ylidene]2-
Pt(CxCPh)2 (2b) was obtained by recrystallization from
CH2Cl2/CH3OH. Yield: 1.218 g (52%) mp: 297–299 8C
(dec). Anal. Calcd for C56 H46 N4 Pt: C, 69.34; H, 4.78; N,
5.78. Found: C, 69.21; H, 4.52; N, 5.44. 1H NMR (300 MHZ,
CDCl3): 1.67 (t, J ¼ 7.2, 2 £ 3H, CH3), 4.73 (q, J ¼ 7.2,
2 £ 2H, CH2), 6.63 (s, 2 £ 2H, CH2An), 6.71 (s, 2 £ 1H,
PhH), 7.21–7.62 (m, 2 £ 10H, imiH,PhH or AnH), 8.06 (d,
J ¼ 8.0, 2 £ 2H, AnH), 8.45 (s, 2 £ 1H, AnH), 8.80 (d,
J ¼ 8.0, 2 £ 2H, AnH), 13C NMR (75 MHz, CDCl3): 16.5
(CH3), 45.9 (NCH2CH3), 57.5 (NCH2An), 79.9 (CxCPh),
112.9 (PtCxC), 119.5 and123.5, 124.8, 125.6, 126.4, 126.6,
127.6, 128.1, 128.5, 129.2, 131.5, 131.8 (AnC, PhC or 4,
5-imiC), 171.1 (Ccarbene). IR (KBr; disk): n (CxC) 2095
A solution of 9-octylimidazole (3.965 g, 0.02 mol)
and 9-chloromethylanthracene (4.983 g, 0.02 mol) in 1,
4-dioxane (150 ml) was stirred for 3 days under refluxing,
and a yellow precipitate was formed. The yellow product was
filtred and washed with 1, 4-dioxane. The yellow crystals of
1-(9-anthracenylmethyl)-3-octylimidazolium chloride are
obtained by recrystallization from CH2Cl2/acetone. yield:
8.169 g (91%). mp: 166–168 8C. Anal. Calcd for
C26H31ClN2: C, 76.73; H, 7.68; N, 6.88. Found: C, 76.34;
H, 7.68; N, 6.62. 1H NMR (300 MHZ, CDCl3): 0.87
(t, J ¼ 6.0, 3H, CH3), 1.25 (m, J ¼ 6.0, 10H, CH2), 1.85
(m, J ¼ 6.0, 2H, CH2), 4.29 (t, J ¼ 6.0, 2H, CH2), 6.68(s, 2H,
CH2An) (An: anthracene), 6.94 (s, 1H, AnH), 7.00 (s, 1H, 4
or 5-imiH) (imi: imidazole), 7.57 (t, J ¼ 8.0, 2H, AnH), 7.65
(t, J ¼ 8.0, 2H, AnH), 8.10 (d, J ¼ 8.0, 2H, AnH), 8.38
(d, J ¼ 8.0, 2H, AnH), 8.61 (s, 1H, 4 or 5-imiH), 11.45 (s, 1H,
2-imiH). 13C NMR (75 MHz, CDCl3): 14.1 (CH3),
22.5(CH2), 26.2 (CH2), 28.9 (CH2), 29.0 (CH2), 30.2(CH2),
31.6 (CH2), 45.5 (CCH2N), 50.2 (AnCH2N), 121.1, 121.5,
122.1 and 122.8, (AnC or 4, 5-imiC), 125.6, 128.3, 129.5,
130.5, 130.9 and 131.2 (AnC), 137.5 (2-imiC).
(m) cm21
.
2.3. [1-(9-Anthracylmethyl)-3-octylimidazol-2-
ylidene]2Pt(CxCPh)2 (2a)
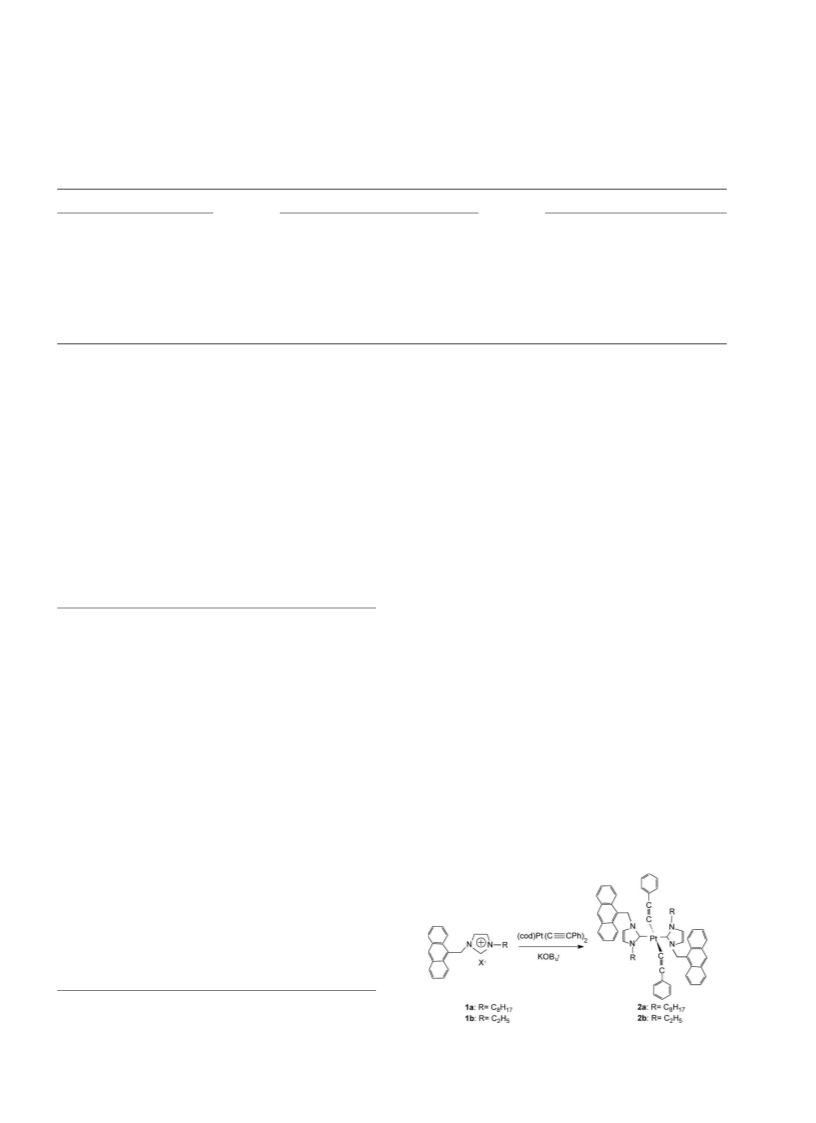
3. Structure determination
For complex 2a, a selected single crystal was mounted on
a Bruker SMART 1000 CCD diffractometer operating at
˚
50 kV and 20 mA using Mo Ka radiation (0.71073 A). Data
A stirred CH3CN and THF (30 ml) suspension of KOBut
(0.090 g, 0.80 mmol), 1, 3-(9-anthracylmethyl)octylimida-
zolium chloride (0.200 g, 0.49 mmol) and (cod)Pt(CxCPh)2
(0.124 g, 0.25 mmol) was refluxed for 8 h, during which
time a brown solution was formed. The solvents were
removed in vacuo, and H2O (30 ml) was added to the
residue, then the solution was extracted with CH2Cl2
(3 £ 20 ml), and the extracting solution was dried with
anhydrous MgSO4, then concentrated to 15 and 3 ml hexane
was added, a pale yellow powder 2a was formed. Yield:
0.163 g (54%). mp: 207–209 8C. Anal. Calcd for C68 H70
N4 Pt: C, 71.75; H, 6.20; N, 4.92. Found: C, 71.48; H, 6.11;
collection and reduction were performed using the SMART
and SAINT software [12] with frames of 0.68 oscillation in
the u range 1:8 , u , 258: An empirical absorption
correction was applied using the SADABS program [13].
The structures were solved by direct methods and all non-
hydrogen atoms were subjected to anisotropic refinement by
full-matrix least squares on F2 using the SHELXTL package
[14]. All hydrogen atoms were generated geometrically
˚
(C–H bond lengths fixed at 0.96 A), assigned appropriated
isotropic thermal parameters and included in structure factor
˚
calculations. Selected bond lengths (A) and angles (8) and
1
N, 4.76. H NMR (300 MHZ, CDCl3): 0.61 (t, J ¼ 7.2,
2 £ 3H, CH3), 1.48 (m, J ¼ 7.2, 2 £ 10H, CH2), 2.17
(m, 2 £ 2H, CH2), 4.61 (t, J ¼ 7.2, 2 £ 2H, CH2), 6.23
(s, 2 £ 2H, CH2An), 6.69 (s, 2 £ 1H, PhH), 7.15–7.46
crystal data and structure refinement for 2a are presented in
Tables 1 and 2, respectively.

 Liu, Qing-Xiang
Liu, Qing-Xiang