Journal of the American Chemical Society
Page 2 of 11
invasive techniques to deliver the complex across cell
membranes.
Na2SO4. Solvent removal under reduced pressure yielded dark
1
2
3
4
5
6
7
8
colored oil. Flash chromatography on alumina using gradient
elution (Et2O→CH2Cl2→CH2Cl2/CH3OH, 9.5:0.5) yielded 5 as
dark brown oil (0.030 g, 45%). TLC (Rf = 0.7, silica, 1:1, CH2Cl2/
CH3OH).1H NMR (500 MHz, CDCl3/ TMS) δ 8.50 (dq, = 4.8, 1.2,
0.8 Hz, 2 H), 8.36 (d, = 2.4 Hz, 1 H), 8.33 (dd, = 4.7, 1.6 Hz, 1 H),
7.87 (dd, = 5.5, 2.2 Hz, 1 H), 7.73 (td, = 8.8, 1.5 Hz, 1 H), 7.64 (td,
= 8.6, 1.7 Hz, 2 H), 7.51- 7.48 (m, 4 H), 7.38 (td, = 8.0, 0.9 Hz, 1
H), 7.12 (td, = 6.7, 1.1 Hz, 2 H), 4.88 (s, 1 H), 4.08 (d, = 14.5 Hz,
2 H), 3.92 (d, =14.6 Hz, 2 H), 3.79 (s, 3 H). 13C NMR (125 MHz)
δ 177.0, 172.1, 159.3, 156.0, 149.0, 137.0, 135.8, 135.1, 132.5,
127.1, 126.9, 124.3, 123.3, 122.4, 122.0, 121.7, 118.6, 118.2,
67.3, 56.9, 52.2. FT-IR (neat, cm-1) 1734.4, 1657.0, 1652.4,
1616.6, 1609.3, 1589.9, 1488.1, 1464.4, 1432.8, 1322.6, 1233.3,
1207.9, 1143.1, 1108.9, 1092.0, 1046.8, 995.2, 882.5, 827.8,
756.1, 725.1, 682.2. HRMS (+ESI) calculated for (C28H24N3O4)
466.1733, observed 466.1761.
EXPERIMENTAL
General synthetic procedures. All materials were obtained
in their highest pure form available from Fisher, Acros Organic
or Alfa Aesar and used without further purification.
Dichloroethane (DCE), dichloromethane (CH2Cl2), acetonitrile
(CH3CN) were purged with argon and dried using a Seca
Solvent Purification System. All chromatography and thin-layer
chromatography (TLC) were performed on silica gel (230-400
mesh) from Silicycle. TLCs were developed using mixtures of
ether/hexanes (Et2O), CH2Cl2/hexanes, ethyl acetate (EtOAc)
/hexanes and CH2Cl2/methanol (CH3OH) as specified and were
visualized with 254 or 365 nm light or stained with iodine (I2).
1H and 13C NMR were recorded using a 500 MHz Bruker-
Biospin NMR instrument, and chemical shifts are reported in
ppm on the δ scale relative to tetramethylsilane (TMS). FT-IR
spectra were recorded using Bruker Optics Vertex 70 with MIR
source as neat crystalline powdered samples and Spectrum
100 Version 10.4.2 (PerkinElmer) fitted with diamond ATR as
oils. LC/MS was carried on a Single Quadruple, Agilent
Technologies 1200 series LC system. High resolution mass
spectra were obtained at the University of Notre Dame mass
spectrometry facility using microTOF instrument operating in
positive ionization mode (+ESI-TOF). Melting-point
information was obtained using Hydrothermal Mel-Temp
instrument. Xantone acetic acid ester (3, XAA-ester) was
prepared as previously described.14
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
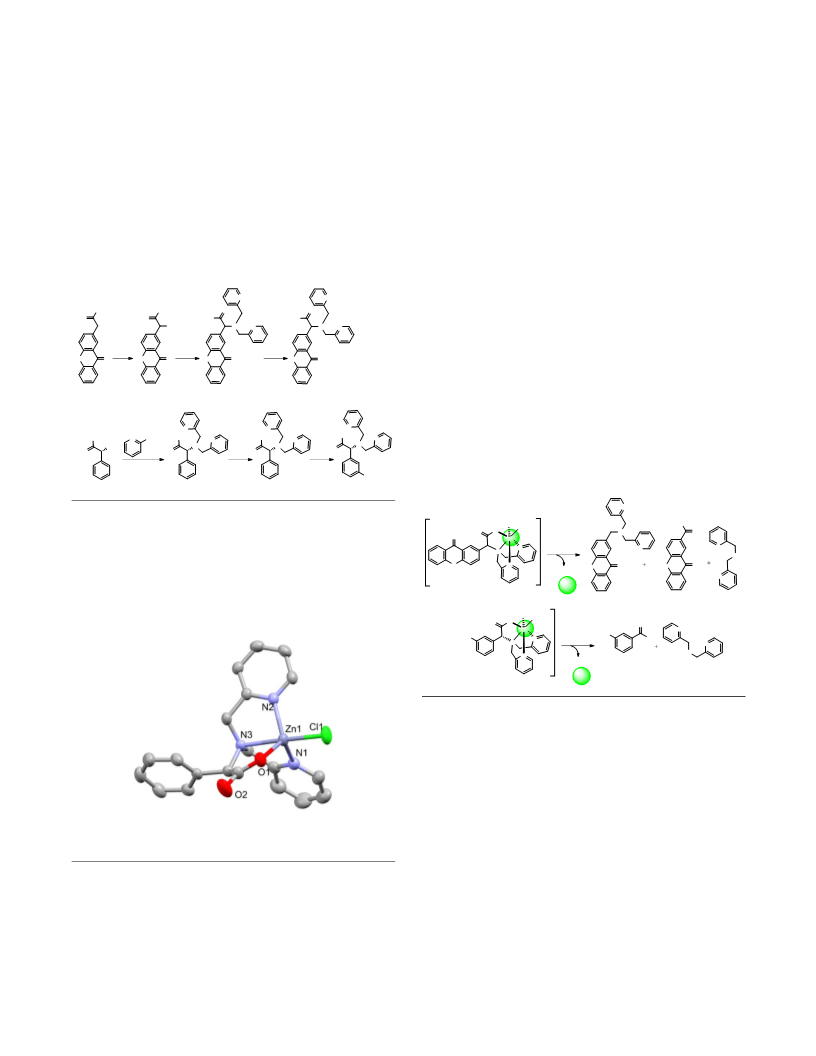
{Bis[(2-pyridyl)methyl]amino}(9-oxo-2-
xanthenyl)acetic acid (XDPAdeCage, 1). Compound 5 (0.15
g, 0.43 mmol) was dissolved in CH3OH (5 mL) and NaOH (0.17
g, 4.3 mmol) was added in small portions. Water (0.5 mL) was
added and the reaction mixture was stirred at room
temperature for 18 h. After removing CH3OH under reduced
pressure, 1 g of crushed ice was added to the resulting semi-
solid material. After adjusting the pH to 6 using HCl, the
product was extracted into EtOAc (2×10 mL). The combined
extracts were dried over Na2SO4. Solvent removal under
reduced pressure yielded 5 as a pale brown solid (0.10 g, 69%).
TLC (Rf = 0.2, silica, 9:1, CH2Cl2/CH3OH); mp = 170-172 °C. 1H
NMR (500 MHz, D2O) δ 8.11 (d, J = 4.4 Hz, 1 H), 7.94 (s, 1 H),
7.75 (dd, J = 9.0, 1.6 Hz, 1 H), 7.49 (td, J = 7.7, 1.2 Hz, 2 H), 7.42
(d, J = 7.3 Hz, 1 H), 7.33 (d, J =7.8 Hz, 2 H), 7.19 (t, J = 7.4 Hz, 1
H), 6.99 (t, J = 6.1 Hz, 2 H), 6.84 – 6.79 (m, 2 H), 6.66 (d, J = 8.4
Hz, 1 H), 4.40 (s, 1 H), 3.79 (dd, J = 16.8, 4.4 Hz, 4 H) 13C NMR
(125 MHz) δ 177.4, 156.0, 154.7, 154.6, 145.2, 140.2, 135.9,
135.4, 134.0, 125.6, 125.0, 124.5, 123.9, 123.6, 119.7, 119.5,
118.2, 117.2, 73.7, 56.7. FT-IR (neat, cm-1) 3063.9, 2837.6,
2325.3, 2161.3, 2051.5, 1979.9, 1706.8, 1656.9, 1610.8, 1592.7,
1570.4, 1489.3, 1464.9, 1434.9, 1368.3, 1349.9, 1322.4, 1269.8,
1221.2, 1140.8, 1123.6, 1095.2, 1051.0, 1019.6, 995.7, 976.1,
964.7, 916.6, 901.3, 883.3, 842.9, 820.5, 762.8, 733.0, 724.6,
682.2, 660.7, 644.6, 632.4, 610.4. HRMS (+ESI) calculated for
(C27H22N3O4), 452.1605 and observed 452.1604.
Methyl bromo(9-oxo-2-xanthenyl)acetate (4). XAA-ester
(1.0 g, 3.7 mmol) was combined with N-bromosuccinimide
(NBS, 0.98 g, 5.5 mmol) and 2,2’- azobisbutyronitrile (AIBN,
0.06 g, 0.37 mmol) in DCE (10 mL). The resulting mixture was
refluxed for 3 h. DCE was removed under reduced pressure and
the product was extracted into CH2Cl2 (2×50 mL), and the
combined organic extracts were dried over Na2SO4. Solvent
removal yielded
a
pale yellow semi-solid. Flash
chromatography on silica using gradient elution (Et2O/hexanes
1:1→Et2O) yielded 4 as pale yellow crystalline solid (0.90 g,
73%). TLC (Rf = 0.5, silica, 1:1, ether/hexanes); mp = 120-
122°C. 1H NMR (500 MHz, CDCl3/ TMS) δ 8.39 (s, 1 H), 8.34 (dd,
J = 7.8, 1.5 Hz, 1 H), 8.06 (dd, J = 8.8, 2.4 Hz, 1 H), 7.75 (td, J =
7.8, 1.5 Hz, 1 H), 7.55 (d, J = 8.8 Hz, 1 H), 7.51 (d, J = 8.7 Hz, 1 H),
7.41 (td, J = 7.5, 0.9 Hz, 1 H), 5.50 (s, 1 H), 3.82 (s, 3 H). 13C NMR
(125 MHz) δ 176.7, 168.7, 156.5, 156.2, 135.5, 135.3, 131.8,
126.9, 124.5, 121.9, 126.9, 124.5, 121.9, 121.6, 119.3, 118.2,
53.7, 45.2. FT-IR (neat, cm-1) 3044.5, 3004.6, 2947.2, 2325.3,
2161.4, 2051.6, 1980.0, 1738.9, 1653.8, 1610.7, 1592.3, 1488.5,
1474.8, 1465.1, 1435.5, 1363.9, 1339.8, 1326.0, 1315.6, 1269.8,
1233.4, 1216.2, 1191.5, 1176.5, 1162.4, 1145.9, 1121.9, 1109.8,
1008.9, 984.0, 926.9, 897.3, 883.8, 864.1, 839.8, 833.4, 807.7,
801.0, 788.9, 761.2, 753.9, 736.0, 724.5, 713.6, 705.1, 676.0,
660.2, 631.1, 603.7. HRMS (+ESI) calculated for (C16H12BrO4)
346.9891, observed 346.9913.
Methyl
{bis[(2-pyridyl)methyl]amino}phenylacetate
(7). Methyl (2S) aminophenyl ethanoate (6, 2.03 g, 12.0 mmol)
was combined with pyridine-2-carboxaldehyde (2.94 g, 27.0
mmol) in DCE (50 mL). NaBH(O2CH3)3 (5.60 mg, 24.4 mmol)
was added in small portions. After 24 h reaction at room
temperature, the reaction mixture was diluted with saturated
NaHCO3 (20 mL). The product was extracted into CH2Cl2 (3×25
mL). The combined extracts were dried over Na2SO4. Solvent
removal under reduced pressure yielded brown oil. Flash
chromatography on silica using gradient elution
(Et2O→CH2Cl2) yielded 7 as white solid (1.25 g, 29.3%). TLC (Rf
1
= 0.5, silica, 9:1, CH2Cl2/CH3OH); mp= 67–69°C. H NMR (500
Methyl
{bis[(2-pyridyl)methyl]amino}(9-oxo-2-
MHz, CDCl3) δ 8.49 (d, J = 4.9 Hz, 2 H), 7.62 (td, J = 7.6, 1.8 Hz, 2
H), 7.50 (d, J = 7.9 Hz, 2 H), 7.41 – 7.39 (m, 2 H), 7.36 – 7.32 (m,
2 H), 7.31 – 7.27 (m, 1 H),7.11 (t, J = 6.7 Hz, 2 H), 4.74 (s, 1 H),
4.07 (d, J = 14.5 Hz, 2 H), 3.89 (d, J = 15.2 Hz, 2 H), 3.75 (s, 3
H).13C NMR (125 MHz, CDCl3) δ 172.7, 160.0, 149.1, 136.5,
136.2, 129.1, 128.6, 128.2, 123.1, 122.1, 67.6, 57.0, 51.9. FT-IR
(neat, cm-1) 3062.0, 3011.2, 2950.4, 2906.5, 2854.9, 2325.4,
2161.2, 2051.5, 1975.2, 1732.9, 1590.0, 1570.6, 1495.5, 1473.5,
xanthenyl)acetate (5). Compound 4 (0.050 g, 0.14 mmol) was
combined with potassium carbonate (K2CO3, 0.0172 g, 0.12
mmol), sodium iodide (0.0187 g, 0.12 mmol), and
dipicolylamine (DPA, 0.025 g, 0.12 mmol) in CH3CN (5 mL).
After 18 h reaction at room temperature, the reaction mixture
was diluted with H2O (10 mL) and the product was extracted
into CH2Cl2 (2×25 mL). The combined extracts were dried over
2
ACS Paragon Plus Environment

 Basa, Prem N.
Basa, Prem N.