Organometallics
Article
Complex 2. To a solution of bpnp (19.9 mg, 0.07 mmol) in DMF
(1 mL) was added Pd(ACN)4(BF4)2 (62.2 mg, 0.14 mmol) under
stirring at room temperature. The reaction mixture was stirred at room
temperature for 5 min. After completion of the reaction, the solvent
was completely removed under reduced pressure and the residue was
washed with acetone and methanol. Upon drying under vacuum, the
pure complex 2 was obtained as a brown-yellow powder (60.8 mg,
ASSOCIATED CONTENT
* Supporting Information
The Supporting Information is available free of charge on the
■
S
1H and 13C NMR spectral data for organic products of
catalysis and bond distances and angles for complex 2
1
0.066 mmol, 94%). H NMR (400 MHz, DMF-d7): δ 9.62 (d, J = 8.7
Hz, 2H), 9.34 (d, J = 8.7 Hz, 2H), 9.11 (d, J = 8.1 Hz, 2H), 8.75−8.71
(m, 4H), 8.17−8.14 (m, 2H). 13C NMR (100 MHz): δ 164.44, 162.57,
156.36, 156.03, 149.94, 146.68, 143.10, 129.71, 128.67, 126.49, 123.97,
35.71, 30.58. Recrystallization of 2 from DMF/methanol at room
temperature gave crystals suitable for X-ray determination. Thus, the
structure of 2 was further confirmed by crystallography. Anal. Calcd
for [2 + 2 DMF]: C, 33.77; H, 3.87; N, 10.50. Found: C, 33.40; H,
3.55; N, 10.61.
Complex 5. A mixture of 1 (25.4 mg, 0.03 mmol) and KCl (5.0
mg, 0.06 mmol) in predried CH3CN (2 mL) was stirred at room
temperature under N2 for 3 h. The solution was centrifuged and
decanted. The residue was washed with Et2O and dried under vacuum
to provide a yellow solid (18.8 mg, 90%). 1H NMR (400 MHz,
DMSO-d6): δ 9.34 (d, J = 8 Hz, 2H), 9.09 (d, J = 8 Hz, 2H), 8.93−
8.74 (m, 4H), 8.45 (t, J = 8 Hz, 2H), 7.83 (t, J = 7 Hz, 2H). 19F NMR
(375 MHz, DMSO-d6): δ −73.84 (s). 13C NMR (100 MHz, DMSO-
d6): δ 162.7, 156.5, 151.9, 149.9, 145.6, 141.8, 128.8, 128.2, 126.2,
123.6. HRMS (ESI): m/z [M − TFA]+ calcd for C18H13Cl2N4OPd2,
582.8536; found, 582.8590.
Accession Codes
CCDC 1558483 contains the supplementary crystallographic
data for this paper. These data can be obtained free of charge
Crystallographic Data Centre, 12 Union Road, Cambridge CB2
1EZ, UK; fax: +44 1223 336033.
AUTHOR INFORMATION
Corresponding Author
ORCID
Notes
The authors declare no competing financial interest.
■
Complex 6. A mixture of 2 (32.5 mg, 3.5 × 10−2 mmol) and KCl
(13.2 mg, 0.18 mmol) in dimethylformamide (1 mL) was stirred at
room temperature under N2 for 3 h. After removal of dimethylforma-
mide, the residue was washed with water and Et2O to give the desired
product as an orange-yellow solid (21.7 mg, 98%). This complex is
insoluble in most organic solvents, even dmso. HRMS (ESI): m/z [M
− Cl]+ calcd for C18H13Cl2N4OPd2, 582.8536; found, 582.8504.
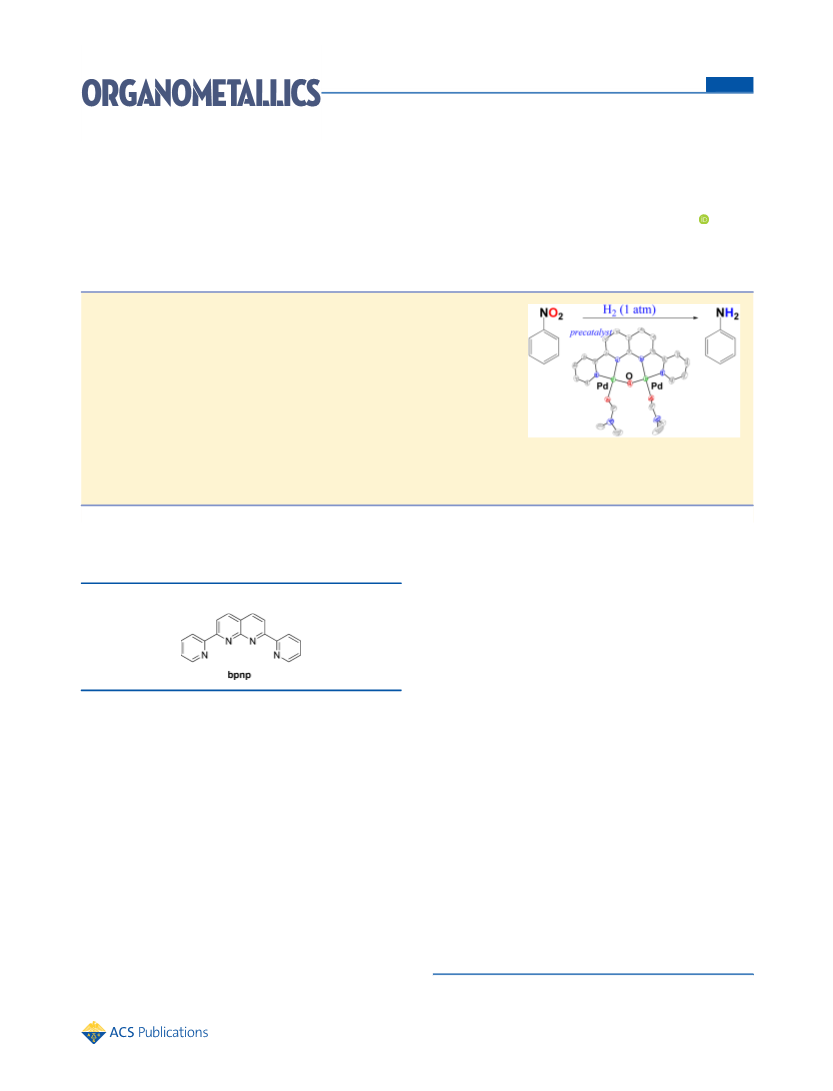
Catalysis Reduction of Nitroarenes. A mixture of nitro
compound (0.5 mmol) and complex 1 (2.5 × 10−3 mmol) in
methanol (0.5 mL) was loaded in a reaction vessel with a stirring bar.
The reaction vessel was flushed with hydrogen gas through an adapter
with a 100 mL balloon filled with H2. The mixture was stirred at 50 °C
for 12 h. After the reaction, methanol was removed under reduced
pressure. The residue was extracted with ether (3 mL × 3), and the
combined organic extracts were dried and concentrated. The residue
was analyzed by NMR spectroscopy. For the purification, chromatog-
raphy on silica gels provided the desired compound in pure form. The
spectral data of the organic products are essentially identical with those
reported. 1H and 13C NMR spectral data for all compounds are
General Kinetic Procedures. A mixture of substrate (1 mmol)
and complex 1 (5 × 10−3 mmol) in methanol (2 mL) was loaded in a
reaction vessel with a stirring bar. The reaction vessel was flushed with
hydrogen gas through an adaptor with a 100 mL balloon filled with H2.
The mixture was stirred at 50 °C. At appropriate time intervals, 0.1 mL
aliquots were removed using a syringe and quickly passed through
Celite to remove the metal complexes with elution of ether. The
filtrate was then concentrated under reduced pressure and analyzed by
1H NMR spectroscopy.
ACKNOWLEDGMENTS
We thank the Ministry of Science and Technology of Taiwan
for financial support (MOST103-2113-M-002-MY3).
■
REFERENCES
■
(1) (a) Tikkanen, W. R.; Krueger, C.; Bomben, K. D.; Jolly, W. L.;
Kaska, W. C.; Ford, P. C. Inorg. Chem. 1984, 23, 3633−3638. (b) Liao,
B.-S.; Liu, Y. H.; Peng, S.-M.; Liu, S.-T. Dalton Trans. 2012, 41, 1158−
1164. (c) Lacroix, P.; Kahn, O.; Valade, L.; Cassoux, P.; Thompson, L.
K. Synth. Met. 1990, 39, 81−90.
(2) (a) Liao, B.-S.; Liu, S.-T. J. Org. Chem. 2012, 77, 6653−6656.
(b) Liao, B.-S.; Liu, S.-T. Catal. Commun. 2013, 32, 28−31. (c) Lan,
Y.-S.; Liao, B.-S.; Liu, Y.-H.; Peng, S.-M.; Liu, S.-T. Eur. J. Org, Chem.
2013, 2013, 5160−5164. (d) Hung, M.-U.; Liao, B.-S.; Liu, Y.-H.;
Peng, S.-M.; Liu, S.-T. Appl. Organomet. Chem. 2014, 28, 661−665.
(e) Bear, J. L.; Chau, L. K.; Chavan, M. Y.; Lefoulon, F.; Thummel, R.
P.; Kadish, K. M. Inorg. Chem. 1986, 25, 1514−1516.
(3) (a) Su, T.-C.; Liu, Y.-H.; Peng, S.-M.; Liu, S.-T. Eur. J. Inorg.
Chem. 2013, 2013, 2362−2367. (b) Tikkanen, W.; Kaska, W. C.;
Moya, S.; Layman, T.; Kane, R.; Krueger, C. Inorg. Chim. Acta 1983,
76, L29−L30.
(4) Binamira-Soriaga, E.; Keder, N. L.; Kaska, W. C. Inorg. Chem.
1990, 29, 3167−3171.
(5) (a) Tikkanen, W. R.; Binamira-Soriaga, E.; Kaska, W. C.; Ford, P.
C. Inorg. Chem. 1984, 23, 141−146. (b) Tikkanen, W. R.; Binamira-
Soriaga, E.; Kaska, W. C.; Ford, P. C. Inorg. Chem. 1983, 22, 1147−
1148.
(6) Yang, F.-M.; Chen, P.-Y.; Lee, C.-C.; Liu, Y.-H.; Peng, S.-M.;
Chou, P.-T.; Liu, S.-T. Dalton Trans. 2012, 41, 5782−5784.
(7) Newkome, G. R.; Garbis, S. J.; Majestic, V. K.; Fronczek, F. R.;
Chiari, G. J. Org. Chem. 1981, 46, 833−839.
Crystallography. A crystal suitable for X-ray determination was
obtained for 2·2DMF. Cell parameters were determined with a
Siemens SMART CCD diffractometer. The structure was solved using
the SHELXS-97 program14 and refined using the SHELXL-97
program15 by full-matrix least-squares on F2 values. Crystal data of
2·2DMF: C30H40B3F12N8O5Pd2, mol wt 1065.93, triclinic, space group
(8) Geary, W. J. Coord. Chem. Rev. 1971, 7, 81−122.
(9) (a) Adrian, R. A.; Broker, G. A.; Tiekink, E. R.T.; Walmsley, J. A.
Inorg. Chim. Acta 2008, 361, 1261−1266. (b) Adrian, R. A.; Benson, R.
E.; Daniels, L. M.; Tiekink, E. R. T.; Walmsley, J. A. Acta Crystallogr.,
Sect. E: Struct. Rep. Online 2006, 62, m601−m603.
(10) Ono, N. The Nitro Group in Organic Synthesis; Wiley-VCH: New
York, 2001.
(11) (a) Jagadeesh, R. V.; Wienhofer, G.; Westerhaus, F. A.; Surkus,
P1; a = 12.7021(4) Å, b = 13.2940(3) Å, c = 13.8549(5) Å, α =
̅
96.541(2)°, β = 103.247(3)°, γ = 93.220(2)°; V = 2254.28(12) Å3; Z
= 2; ρcalcd. = 1.570 Mg m−3; F(000) = 1062; crystal size 0.25 × 0.20 ×
0.20 mm3; 22435 reflections collected; 9983 independent reflections
(R(int) = 0.0384); θ range 3.05−27.50°; goodness of fit on F2 1.067;
final R indices (I > 2σ(I)) R1 = 0.0605, wR2 = 0.1610; R indices (all
data) R1 = 0.0922, wR2 = 0.1821. CCDC 1558483. Other
̈
A.-E.; Junge, H.; Junge, K.; Beller, M. Chem. - Eur. J. 2011, 17, 14375−
F
Organometallics XXXX, XXX, XXX−XXX

 Yang, Shu-Ting
Yang, Shu-Ting