10.1021/jo202119p
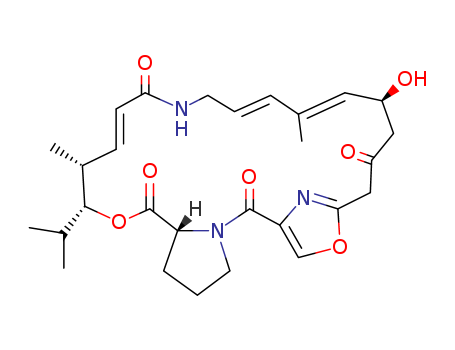
The research focuses on the stereoselective synthesis of the antibiotic (?)-virginiamycin M2. The study aims to develop an efficient and enantioselective synthesis of this complex antibiotic, which is part of the group A virginiamycins known for their potent antibiotic activity against various resistant bacterial strains. The researchers employed a convergent strategy involving a series of key steps, including the use of chiral silane reagents prepared through Rh(II)- or Cu(I)-catalyzed carbenoid Si?H insertion to introduce the desired olefin geometry and stereocenters. They also utilized a modified Negishi cross-coupling or titanium-mediated alkyne?alkyne reductive coupling to assemble the trisubstituted (E,E)-diene and employed a SmI2-mediated macrocyclization to construct the 23-membered macrocycle scaffold. The synthesis was completed with a final deprotection step to reveal (?)-virginiamycin M2. The study concludes that the developed synthetic route is highly convergent, achieving the total synthesis in 19 steps with a longest linear sequence of 10 steps, and offers a modular approach that could facilitate the design and synthesis of analogues for further exploration of the biological potential of group A virginiamycins.