594 JOURNAL OF CHEMICAL RESEARCH 2014
Table 1 Yields of 2a in different reaction conditionsa
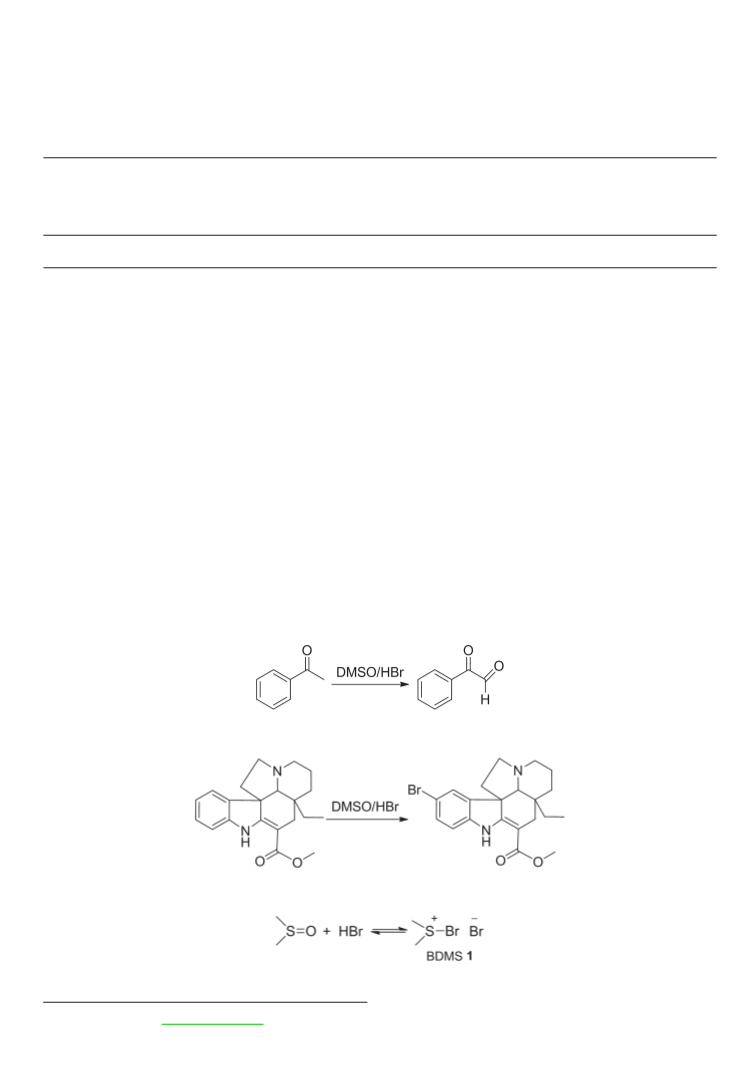
with a yield of 43% after 24 h at room temperature. In the
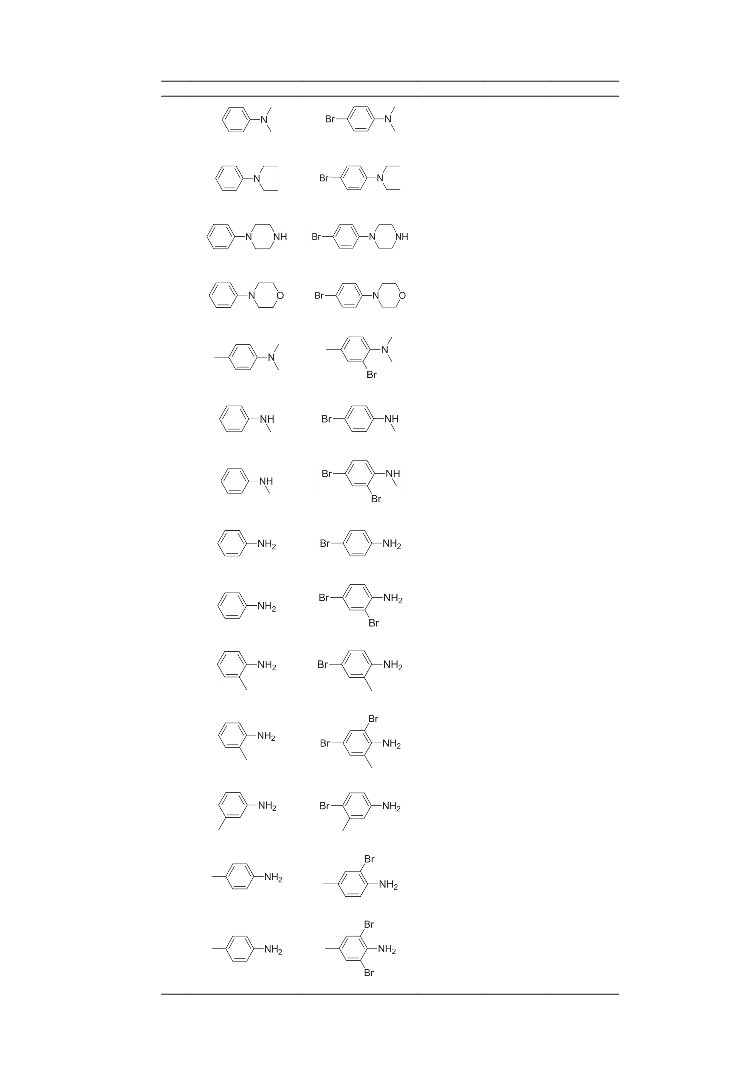
present work, N,N-dimethylaniline was para-monobrominated
in DMSO/HBr system with a yield of 96% at 60 °C (Table 3,
entry 1). Other tertiary amines, such as N,N-diethylaniline,
N-phenylmorpholine and N-phenylpiperazine were also
efficiently para-monobrominated at 60 °C (Table 3, entries
2–4). When the para-position of the tertiary arylamine
was blocked by a methyl group, ortho-monobromination
occurred at 70 °C (Table 3, entry 5). With secondary or
primary amines substrates, mono- or di-bromination could be
achieved by regulating the temperature. At 50 °C, aniline and
N-methylaniline were para-monobrominated; while at 80 °C,
the main reaction was ortho–para-dibromination (Table 3,
entries 6–9). With o-toluidine, monobromination occurred
at the ortho-position at 50 °C whilst the main reaction was
ortho-para-dibromination at 80 °C (Table 3, entries 10–11).
m-Toluidine was also para-brominated at 50 °C (Table 3, entry
12). No obvious dibromination occurred at 80 °C. When the
para-position of aniline was blocked by a methyl group, ortho-
monobromination occurred at 50 °C and ortho-dibromination
occurred at 80 °C (Table 3, entries 13 and 14). The above
results indicate that in the DMSO/HBr system, bromination
of aniline occurred first at the para-position and then at the
ortho-position. Mono- and di-bromination of secondary and
primary amines occurred at 50 °C and 80 °C respectively. To
prove the practicality of this method for larger-scale synthesis,
4-bromo-N,N-dimethylaniline 8a was prepared on a gram scale
(10 mmol) to give a 95% yield of isolated product.
H
O
H
O
°
HBr, 50 C, 2h
N
N
solvent
Br
2a
2
Entry
Solvent
Yield of 2a/%b
1
2
3
4
5
6
DMF
EtOAc
EtOH
acetonitrile
DMSO
0
0
0
0
83
AcOH/DMSO (2:1)
21
aReaction was performed with 1 mmol 2-acetylpyrrole (2) and 1 mL HBr in
1 mL solvent at 50 °C for 2 h.
bIsolated yield.
The successful bromination of 2-acetylpyrrole encouraged
us to expand this reagent to other aza aromatic compounds.
The results are shown in Table 2. Pyrrole-2-carboxaldehyde
and carbazole were also efficiently monobrominated at 50 °C
(Table 2, entries 2 and 3). The addition of an alkyl group into
2-acetylpyrrole mainly induced 4,5-dibromination (Table 2,
entries 4–6), probably because of the activating nature of groups
such as methyl and ethyl that facilitate dibromination.22–24 The
yield of compound 3a gained through the DMSO/HBr system
is higher than those obtained by traditional methods.25,26
According to Majetich et al.20 the DMSO/HBr system was
reported to effect the bromination of N,N-dimethylaniline
Conclusion
We have developed a highly efficient approach for the
bromination of pyrrole derivatives, carbazole and aromatic
amines with high yields. Temperature control used with
primary and secondary amines helped to promote mono- or
di-bromination. The reaction proceeds under mild conditions,
and is amenable to the gram-scale synthesis of brominated
anilines. Simplicity of operation, low toxicity and high
selectivity make it a promising new bromination procedure.
Table 2 Bromination of aza aromatic compounds using DMSO/HBra
Entry
Substrate
Product
T/°C
Time/h Yield/%b
1
50
2
2
83
73
2
2a
Experimental
2
3
50
50
All reagents and solvents were purchased from J&K Chemical Co. and
used without further purification. Melting points were determined on
a SGW X-4 micro melting point instrument. 1H and 13C NMR spectra
were recorded on Varian 400 or Bruker 500 MHz spectrometers. IR
spectra were obtained on a Perkin Elmer FTIR system. HRMS spectra
were obtained using a LTQ Orbitrap Discovery spectrometer from
Thermo Fisher.
3
4
3a
2
87
4a
5b
Synthesis of bromination products: general procedure
In a round-bottomed flask, the substrate (1 mmol) and aqueous HBr
(48%) (1 mL) were mixed in DMSO (1 mL). The mixture was stirred
at corresponding temperature for 1–4 h. After cooling to room
temperature, the reaction was adjusted to pH 7–8 with aqueous NaOH
solution (4 M). Then the mixture was washed twice with EtOAc, and
the combined organic extracts were dried, filtered and concentrated
under reduced pressure to give bromination products.
4
5
50
50
2
2
85
91
5
6
6b
Electronic Supplementary Information
The structure of compound 2a was confirmed by X-ray
crystallography. The details of the crystal data have been
deposited with Cambridge Crystallographic Data Centre as
Supplementary Publication, CCDC 890435. Full details of the
physical and spectroscopic data (1H and 13C NMR and HRMS)
for all the compounds reported in this paper (Tables 2 and 3)
and the X-ray crystal structure of 2a is available as ESI through:
stl.publisher.ingentaconnect.com/content/stl/jcr/supp-data
50
2
83
6
7
7b
aReaction was performed with 1 mmol substrate and 1 mL HBr in 1 mL
DMSO at 50 °C for 2 h.
bIsolated yield.

 Liu, Cong
Liu, Cong