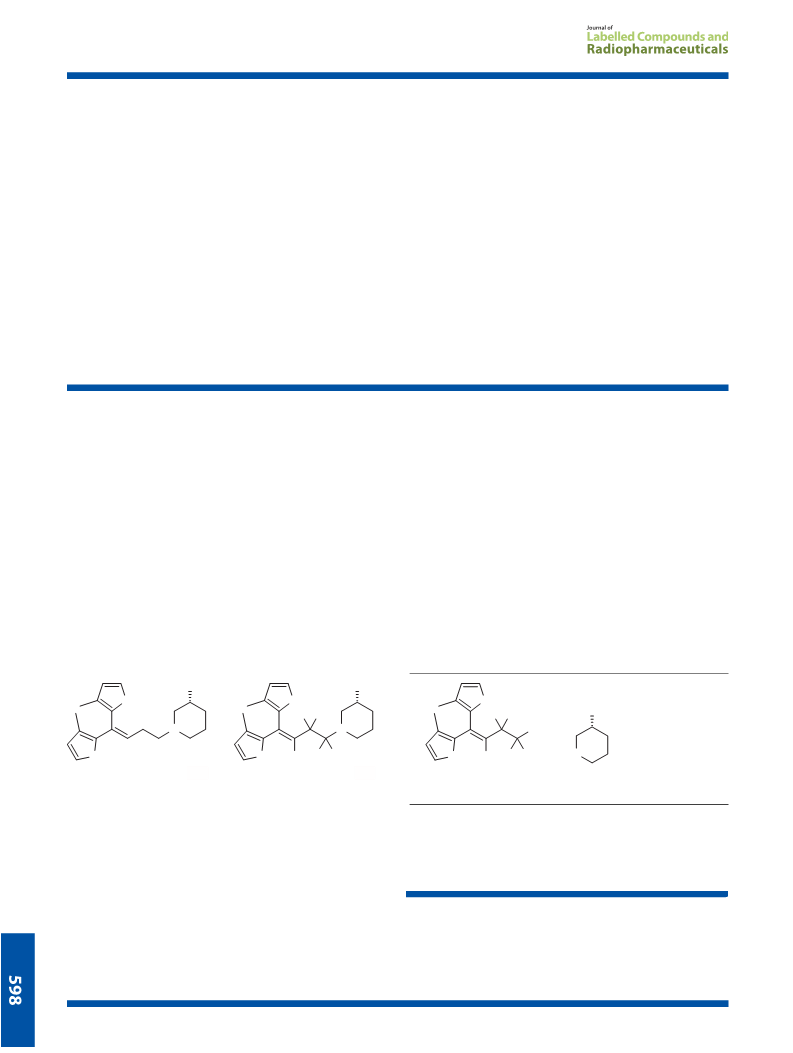
J. M. Herbert and T. W. Mathers
were recorded using a Bruker Micro-TOF instrument. Reagents phase was re-extracted twice with ether, and the combined
were obtained commercially, with [2H6]-g-butyrolactone being organic phases were washed with brine and evaporated to leave
obtained from Isotec Inc. Ethyl R(1)-nipecotate was obtained by an oil, which was redissolved in 2-propanol (15 ml). Aqueous
resolution of racemic material using L-tartaric acid;6 the material sulfuric acid (20%, 1.5ml) was added, and the mixture was stored
obtained by these means had an enantiomeric excess of 91% as for 3 h at room temperature. Volatiles were removed under
1
determined from H n.m.r. of the urea formed upon treatment reduced pressure and the residue was diluted with water and
with 1-(1-naphthyl)ethyl isocyanate.7
extracted three times with dichloromethane. The combined
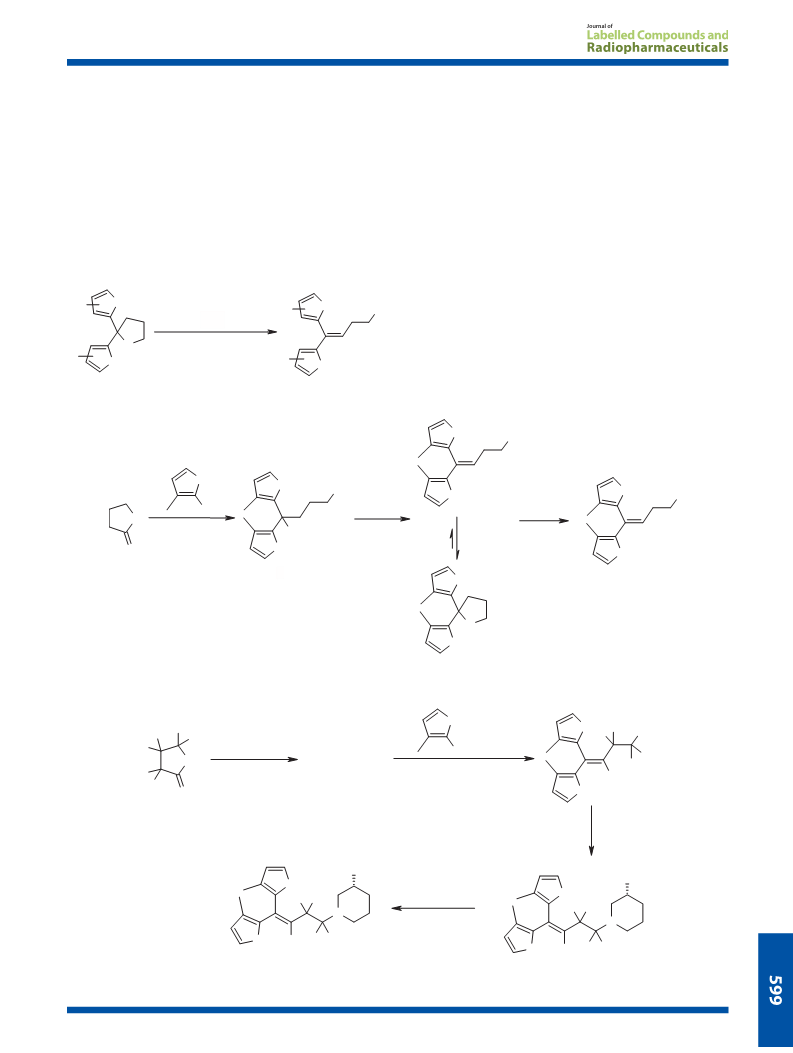
1,1-Bis(3-methyl-2-thienyl)butane-1,4-diol (4). 2-Bromo-3-methyl extracts were washed with saturated aqueous sodium hydro-
thiophene (1.932g, 11 mmol) in ether (15ml) was added under gencarbonate, dried (Na2SO4) and evaporated to leave [butene-
nitrogen at 5–101C to a stirred solution of butyllithium (1.6 M in 2H5]-1,1-bis(3-methyl-2-thienyl)-4-bromo-1-butene as an oil (4.39g;
hexanes: 7.5 ml, 12 mmol) in ether (7.5 ml). The mixture was stirred 70% pure by GC), which was used directly in the next step.
for 15 min, cooled to À601C, and g-butyrolactone (0.330 ml,
4.3mmol) was added. After a further 2.5 h, water (15 ml) was
added followed by 1M hydrochloric acid (15 ml). The organic
phase was separated and the aqueous phase re-extracted with
[butene-2H5]-Ethyl (R)-1-[4,4-bis(3-methyl-2-thienyl)-3-bute-
nyl]-3-piperidinecarboxylate (7)
A suspension of potassium iodide (0.23 g, 1.4 mmol) and potassium
carbonate (2.24 g, 16.2 mmol) in acetone containing 2b (4.394 g,
13.4 mmol) and ethyl R(1)-nipecotate (2.18 g, 13.8 mmol) was
stirred at room temperature for 46 h, then filtered. The filtrate was
evaporated and the residue was chromatographed in 9:1 hexane-
THF on silica gel to give ethyl [butene-2H5]-(R)-1-[4,4-bis(3-methyl-
2-thienyl)-3-butenyl]-3-piperidinecarboxylate (3.367 g, 62% from
methyl [butyric acid-2H5]-4-bromobutyrate). dH(CDCl3) 1.22 (3H, t),
1.3–1.6 (2H, m), 1.68 (1H, m), 1.92 (1H, m), 2.01 (3H, s), 2.02 (3H, s),
2.12 (1H, t), 2.52 (1H, m), 2.71 (1H, m), 2.93 (1H, m), 3.10 (1H, m),
4.19 (2H, q), 6.74 (1H, d), 6.82 (1H, d), 7.02 (1H, d), 7.18 (1H, d). m/z
408 (M1Á ), 172 (100%).
[butene-2H5]-Tiagabine hydrochloride (1b). Aqueous sodium
hydroxide (12 M, 1.5 ml) was added to a solution of 7 (3.367 g,
8.25 mmol) in ethanol (20 ml). The mixture was stirred for 4 h
at room temperature, cooled to 51C, and acidified to pH 1
with 4 M hydrochloric acid. The mixture was extracted with
dichloromethane (400 ml) and the extract washed with water
(5 ml). Solvent was removed under reduced pressure and
the residue was recrystallised from isopropanol to give
[butene-2H5]-tiagabine hydrochloride (2.056 g, 60%), m.p.
182–1851C (lit.2 183.5–185.51C). [a]DÀ201 (C 1, H2O). dH(CD3OD)
1.5–2.3 (4H, m), 1.99 (3H, s), 2.06 (3H, s), 2.7–3.3 (3H, m), 3.3–3.8
(2H, m), 6.80 (1H, d), 6.95 (1H, d), 7.19 (1H, d), 7.39 (1H, d);
dH(CH3OH-CDCl3) 1.99 (2D), 2.60 (2D), 5.40 (1D); m/z (esi-TOF)
381 (MH1, 100%), 282, 252; HRMS found: 381.1718 (calc. for
C20H21 2H5NO2S2 381.1719).
ether. The combined ethereal phases were washed with water and
brine, and dried (MgSO4). Solvent was removed under reduced
pressure to leave 4 (1.025g, 84%) as a white solid. dH(CDCl3) 1.59
(2H, m), 1.85 (6H, s), 2.45 (2H, t), 3.54 (2H, t), 6.76 (2H, d), 7.12
(2H, d); m/z 282 (M1), 264 (M-H2O1), 233, 223, 125 (100%).
Example Procedure: 4,4-bis(3-methyl-2-thienyl)-3-buten-1-ol (5),
and 2,2-bis(3-methyl-2-thienyl)tetrahydrofuran (6). A solution of 4
(304mg, 1.1 mmol) in toluene (30 ml) containing 4-toluenesulfo-
nic acid (22 mg) was heated at reflux for 45min, cooled, and
washed with aqueous sodium hydrogencarbonate. The organic
phase was dried (MgSO4) and solvent was removed under
reduced pressure. Column chromatography of the residue in
ethyl acetate–hexane (1:3) on silica gel gave 6 (130mg, 45%).
dH(CDCl3) 1.97 (6H, s), 2.07 (2H, pentet), 2.60 (2H, t), 4.05 (2H, t),
6.75 (2H, d), 7.04 (2H, d); m/z 264 (M1 Á ), 249, 207 (100%), 167, 125
(100%); HRMS (esi-TOF) 265.0753 (calc. for C14H17OS2 265.0715).
Further elution gave 5 (91 mg, 32%). dH(CDCl3) 2.00 (3H, s), 2.05
(3H, s), 2.41 (2H, appears as q), 3.71 (2H, t), 6.09 (1H, t), 6.74 (1H, d),
6.84 (1H, d), 7.05 (1H, d), 7.20 (1H, d); m/z 264 (M1 Á ), 233, 111
(100%); HRMS (esi-TOF) 265.0730 (calc. for C14H17OS2 265.0715).
Methyl [butyric acid-2H6]-4-bromobutyrate. Boron tribromide in
dichloromethane (1 M; 65ml) was added to a stirred solution of
[2H6]-g-butyrolactone (5.0g, 54mmol) in dichloromethane (60 ml)
under nitrogen at room temperature. The mixture was stirred for
18h, following which additional boron tribromide in dichlor-
omethane (1M; 15ml) was added. After a further 3 h, anhydrous
methanol (200ml) and additional dichloromethane (125ml) were
added. The mixture was stirred for a further 10min, and then
washed with aqueous sodium carbonate, aqueous sodium
thiosulfate, and water (200 ml each). The organic phase was
dried (MgSO4) and solvents were removed under reduced
pressure to leave methyl [butyric acid-2H6]-4-bromobutyrate
(6.636g, 66%) as a colourless liquid. dH(CDCl3) 3.68 (s); dH(CHCl3)
2.14, 2.49, 3.44; m/z 186, 188 (M1 Á ), 155, 157 (M-ÁOMe), 127/129
(155-CO), 77 (CD3COOMe1 Á , 100%); HRMS(NH3-CI1) Found:
204.0505 (calc. for C5H3 2H6BrO2.NH41 204.0506).
Acknowledgements
The authors thank Dr. S. J. Byard for NMR spectra, and Dr. T. Dransfield
(University of York) for high-resolution mass spectra.
References
[butene-2H5]-1,1-Bis(3-methyl-2-thienyl)-4-bromo-1-butene (2b)
[1] M. H. Pollack, P. P. Roy-Byrne, M. van Ameringen, H. Snyder,
C. Brown, J. Ondrasik, K. Rickels, J. Clin. Psychiatry 2006, 66, 1401.
[2] K. E. Andersen, C. Braestrup, F. C. Grønwald, A. S. Jørgensen,
E. B. Nielsen, U. Sonnewald, P. O. Sørensen, P. D. Suzdak,
L. J. S. Knutsen, J. Med. Chem. 1993, 36, 1716.
Butyllithium in hexanes (1.6 M; 20 ml) was added under nitrogen at
5–101C to a stirred solution of 2-bromo-3-methylthiophene
(5.66 g, 32 mmol) in ether (30 ml). The mixture was stirred for
15 min, and then cooled to À701C and methyl [butyric acid-2H6]-4-
bromobutyrate (2.504g, 13mmol) was added. After stirring for a
further 3 h at the same temperature, the mixture was quenched
by addition of aqueous ammonium chloride and allowed to warm
to room temperature. The phases were separated, the aqueous
[3] J. F. Vozza, J. Org. Chem. 1959, 24, 720.
[4] U. Sonnewald, Acta Chem. Scand. 1988, 42(B), 567.
[5] G. A. Olah, R. Karpeles, S. C. Narang, Synthesis 1982, 963.
[6] A. M. Akkerman, D. K. De Jongh, H. Veldstra, Rec. Trav. Chim.
Pays-Bas 1951, 70, 899.
[7] P. Magnus, L. S. Thurston, J. Org. Chem. 1991, 56, 1166.
View this article online at
wileyonlinelibrary.com
Copyright r 2010 John Wiley & Sons, Ltd.
J. Label Compd. Radiopharm 2010, 53 598–600

 Herberta, John M.
Herberta, John M.