7
608
P. M. Pihko, A. Erkkil a¨ / Tetrahedron Letters 44 (2003) 7607–7609
13
12
have been extensively studied by Evans and others.
Kontturi and Ari Koskinen (both at HUT) for access
to chiral GC and for material support, Dr. Jari
Koivisto (HUT) for NMR assistance, and Dr. Esko
Karvinen (Perstorp/Dynea Chemicals) for a gracious
gift of aldehydes 4 and 5.
However, to the best of our knowledge, diastereoselec-
tive aldol reactions between chiral a,b-disubstituted
aldehydes and ketene silyl acetals derived from esters
have not been previously documented. To our delight,
exposure of aldehyde 7 to ketene silyl acetal 3 in the
presence of BF ·Et O afforded the desired Felkin
3
2
product
2
in 65% yield as the only observed
References
1
4
diastereomer. This result thus supports the Evans
1
2
model.
1. (a) Notz, W.; List, B. J. Am. Chem. Soc. 2000, 122,
7
386–7387; (b) List, B.; Lerner, R. A.; Barbas, C. F. J.
With the carbon skeleton of prelactone B in place, final
Am. Chem. Soc. 2000, 122, 2395–2396; (c) List, B.; Pojar-
liev, P.; Castello, C. Org. Lett. 2001, 3, 573–575; (d) List,
B. Tetrahedron 2002, 58, 5573–5590; (e) List, B. Synlett
global deprotection and lactonization with HF in
1
5
MeCN/H O gave (−)-prelactone B 1 as a highly crys-
2
talline solid (mp 98–99°C). Synthetic 1 proved to be
identical to the natural product in all respects (mp, IR,
NMR, HRMS) with the exception of its optical rota-
2
001, 11, 1675–1686; (f) Pidathala, C.; Hoang, L.; Vig-
nola, N.; List, B. Angew. Chem., Int. Ed. 2003, 42,
785–2788.
1
6
2
tion, which was similar in magnitude but opposite in
2
. (a) Sakthivel, K.; Notz, W.; Bui, T.; Barbas, C. F., III J.
Am. Chem. Soc. 2001, 123, 5260–5267; (b) C o´ rdova, A.;
Notz, W.; Barbas, C. F., III J. Org. Chem. 2002, 67,
7
sign ([h] =−46 (c 0.42, MeOH); lit. [h] =+38.3 (c 0.6,
D
D
MeOH). Thus, the unnatural enantiomer of 1 was
produced; the natural (+)-1 enantiomer could similarly
3
01–303; (c) Chowdari, N. S.; Ramachary, D. B.; C o´ r-
dova, A.; Barbas, C. F., III Tetrahedron Lett. 2002, 43,
591–9595; (d) C o´ rdova, A.; Notz, W.; Barbas, C. F., III
be accessed starting with
D-proline as the catalyst.
9
In summary, we have achieved a short, catalytic, enan-
tioselective and nearly completely diastereoselective
synthesis of (−)-prelactone B in only four steps and 22%
overall yield. Our synthesis is the shortest reported so
Chem. Commun. 2002, 1, 3024–3025.
. Northrup, A. B.; MacMillan, D. W. C. J. Am. Chem.
Soc. 2002, 124, 6798–6799.
. (a) Kumaragurubaran, N.; Juhl, K.; Zhuang, W.;
Bøgevig, A.; Jørgensen, K. A. J. Am. Chem. Soc. 2002,
3
4
17
far and requires only inexpensive reagents. Studies to
combine the proline-catalyzed aldol reactions with the
realm of aqueous Mukaiyama aldol chemistry are in
progress.
124, 6254–6255; (b) Bøgevig, A.; Kumaragurubaran, N.;
Jørgensen, K. A. Chem. Commun. 2002, 6, 620–621.
. The intramolecular version of the proline-catalyzed
ketone–ketone aldol reaction is known as the Hajos–Par-
rish–Eder–Sauer–Wiechert process: (a) Hajos, Z. G.; Par-
rish, D. R. J. Org. Chem. 1973, 38, 3239–3243; (b) Hajos,
Z. G.; Parrish, D. R. J. Org. Chem. 1974, 39, 1615–1621
and references cited therein; (c) Eder, U.; Sauer, G.;
Wiechert, R. Angew. Chem., Int. Ed. 1971, 40, 496-497.
Intramolecular proline-catalyzed aldehyde–ketone aldol
reactions date back to the Woodward synthesis of ery-
thromycin: (d) Woodward, R. B.; Logusch, E.; Nambiar,
K. P.; Sakan, K.; Ward, D. E.; Au-Yeung, B.-W.; Bal-
aram, P.; Browne, L. J.; Card, P. J.; Chen, C. H.;
5
Acknowledgements
Financial support from Hormos Medical Ltd and HUT
is gratefully acknowledged. We thank Professors Ky o¨ sti
Ch e/ nevert, R. B.; Fliri, A.; Frobel, K.; Gais, H.-J.;
Garratt, D. G.; Hayakawa, K.; Heggie, W.; Hesson, D.
P.; Hoppe, D.; Hoppe, I.; Hyatt, J. A.; Ikeda, D.; Jacobi,
P. A.; Kim, K. S.; Kobuke, Y.; Kojima, K.; Krowicki,
K.; Lee, V. J.; Lautert, T.; Malchenko, S.; Martens, J.;
Matthews, R. S.; Ong, B. S.; Press, J. B.; Rajan Babu, T.
V.; Rousseau, G.; Sauter, H. M.; Suzuki, M.; Tatsuta,
K.; Tolbert, L. M.; Truesdale, E. A.; Uchida, I.; Ueda,
Y.; Uyehara, T.; Vasella, A. T.; Vladuchick, W. C.;
Wade, P. A.; Williams, R. M.; Wong, H. N.-C. J. Am.
Chem. Soc. 1981, 103, 3210–3213.
6
. Three previous syntheses of prelactone B have been
reported in the literature: (a) Hanefeld, U.; Hooper, A.
M.; Staunton, J. Synthesis 1999, 401–403; (b) Fournier,
L.; Gaudel-Siri, A.; Kocie n´ ski, P. J.; Pons, J.-M. Synlett
2003, 107–111; (c) Chakraborty, T. K.; Tapadar, S. Tet-
rahedron Lett. 2003, 44, 2541–2543. In addition, a concise
synthesis of related prelactone C was described recently:
(d) Yamashita, Y.; Saito, S.; Ishitani, H.; Kobayashi, S.
J. Am. Chem. Soc. 2003, 125, 3793–3798.
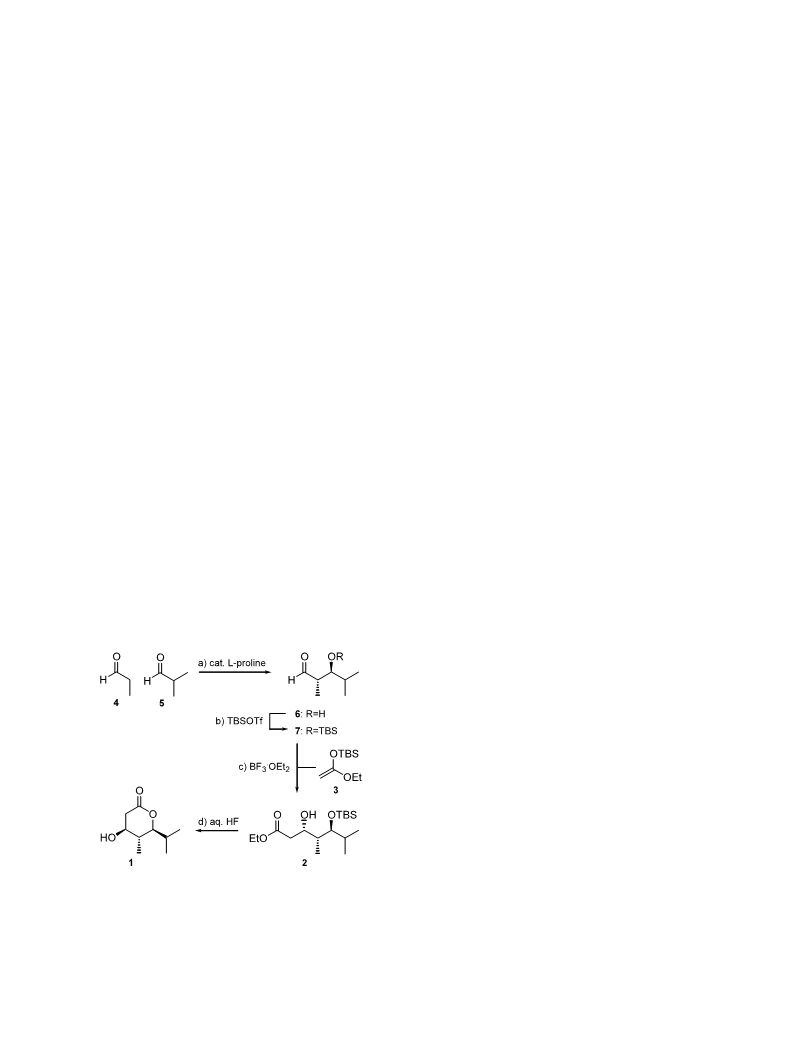
Scheme 2. Reagents and conditions: (a) 5 (400 mol%),
line (10 mol%), DMF, addition of 4 by syringe pump over 30
h; then 10 h, 5°C; (b) crude 6 (dried over 4 A MS), TBSOTf
170 mol%), 2,6-lutidine (330 mol%), 1:1 Et O/CH Cl , −20 to
L-pro-
,
(
2
2
2
1
0°C, 2.5 h, 61% (two steps); (c) BF ·Et O (100 mol%), 3 (300
3 2
mol%), CH Cl , −78°C, 65%; (d) 48% HF, H O, MeCN
2
2
2
(
1:2:17), 4.5 h, rt, 55%.

 Pihko, Petri M.
Pihko, Petri M.