10.1021/ja000108e
The research focuses on a novel synthesis method for β-lactam compounds, which are important in pharmaceutical applications. The purpose of the study was to explore the behavior of spirocyclopropane isoxazolidines under different conditions, specifically with strong acids, to develop a new route for synthesizing valuable β-lactam derivatives. The researchers used readily available and inexpensive anthranilic acid as a starting material, employing tosyl (Ts) and nosyl (2-nitrobenzenesulfonyl, Ns) groups to protect the amino acid nitrogen atom. The process involved the preparation of alkylidenecyclopropanes through palladium(0) catalysis, followed by conversion to isoxazolidines and subsequent heating in the presence of trifluoroacetic acid (TFA) or other strong acids.
10.1016/j.jfluchem.2011.11.005
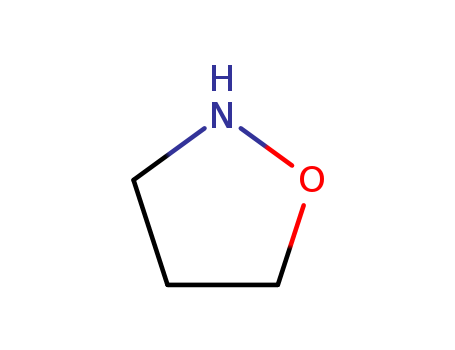
The research aims to develop a new class of fluorinated isoxazolidine derivatives of pyrimidine through a two-step synthesis process involving Suzuki–Miyaura coupling followed by 1,3-dipolar cycloaddition with nitrones. The purpose is to explore the synthesis of fluorinated analogues of biologically important compounds, which have unique properties relevant to medicinal chemistry and biochemistry. Key chemicals used include 5-(dihydroxyboryl)-2,4-bis(alkoxy)-pyrimidines, various fluorovinyl halides such as 1,1,2-trifluoro-2-iodoethene and perfluoroprop-1-enyl halides, and nitrones generated from paraformaldehyde and substituted hydroxylamines. The study concludes that the fluorinated isoxazolidines were successfully synthesized with complete regioselectivity and a good degree of diastereoselectivity. Mild hydrolysis conditions allowed for the removal of pyrimidine protecting groups without breaking the N–O bond of the isoxazolidine ring, leading to a fluorinated analogue of pseudouridine. The results suggest that this method provides a viable route for synthesizing fluorinated nucleoside analogues with potential applications in medicinal chemistry.


 Xi
Xi





