Refernces
10.1021/ol0157813
The study presents a novel strategy for synthesizing polypeptides using recombinant proteins, which are nonprotected peptides, in conjunction with S-alkyl peptide thioesters as building blocks. The method involves oxidizing the N-terminal serine of a peptide to form an Nr-glyoxyloyl peptide, which then undergoes reductive amination with 4,5-dimethoxy-2-(triphenylmethylthio)benzylamine to attach a thiol linker. This results in an Nr-4,5-dimethoxy-2-mercaptobenzyl glycyl peptide, which can be condensed with a peptide thioester to form a peptide bond. The innovative aspect of this approach is the use of the 4,5-dimethoxy-2-mercaptobenzyl (Dmmb) group as a linker, which can be removed under acidic conditions, allowing for the synthesis of peptides with native peptide bonds. The study demonstrates this method using a model sequence and shows the successful preparation of a thiol linker-attached peptide for condensation with peptide thioesters, providing a useful method for peptide synthesis in a neutral aqueous environment without the need for protecting groups.
10.1039/c3ob41936c
The research presented in the "Organic & Biomolecular Chemistry" paper focuses on the discovery of potential anti-inflammatory drugs, specifically diaryl-1,2,4-triazoles bearing an N-hydroxyurea moiety, which serve as dual inhibitors of cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX). The study involves the synthesis and evaluation of a series of hybrid compounds derived from diaryl-1,2,4-triazole and hydroxamic acid or N-hydroxyurea, designed to act as novel anti-inflammatory agents. The synthesized compounds were biologically tested for their inhibitory activities against COX-2 and 5-LOX in vitro, with compound 15e showing optimal inhibitory activities. The selectivity of these compounds for COX-2 over COX-1 was also evaluated, with 15e demonstrating a selectivity index comparable to celecoxib. Additionally, the anti-inflammatory activity of selected compounds was assessed using xylene-induced ear edema in mice, albumen-induced paw edema in rats, and acetic acid-induced vascular permeability in mice models. The analgesic activity was evaluated using acetic acid-induced writhing response and hot-plate assays. Molecular modeling studies were conducted to understand the binding interactions of compound 15e with COX-2 and 5-LOX. The research suggests that compound 15e may be a promising anti-inflammatory agent for further evaluation. The reactants used in the synthesis include para-position substituted phenylhydrazine hydrochloride, ethyl 3-bromopropionate, hydroxylamine methanol solution, KOH, and various substituted phenyl rings, among others. The analyses involved high-performance liquid chromatography (HPLC), electrospray ionisation (ESI) mass spectrometry, infrared (IR) spectroscopy, and nuclear magnetic resonance (NMR) spectroscopy to determine the structures and purities of the synthesized compounds.
10.1039/b010224p
The research presented in the document focuses on a novel and efficient synthetic approach to 6-amidino-2-oxopurines, which are significant in medicinal chemistry. The study employs the reaction between 5-amino-4-cyanoformimidoylimidazoles and tosyl isocyanate to produce the desired 6-amidino-2-oxopurines with high yields. The reaction's mechanism was explored through additional experiments involving selective acylation of imidazoles and reactivity studies with various compounds. The structures of the synthesized compounds were confirmed using X-ray crystallography, and their properties analyzed through techniques like infrared (IR), nuclear magnetic resonance (NMR), and mass spectrometry. The research also discusses the rearrangement of these compounds in the presence of acetic acid and DMF, leading to the formation of pyrimido[5,4-d]pyrimidin-2-one, with the structural confirmation of this compound through X-ray crystallography. The detailed experimental procedures, including the preparation of starting materials, reaction conditions, and analytical methods, are provided, highlighting the use of elemental analysis, spectroscopic data, and crystallographic studies for a comprehensive understanding of the synthesized compounds and their properties.
10.1007/BF00506677
The research focuses on the isomerization of thiazolo[4,5-b]quinoxalines in the presence of acids, investigating the pathways of their isomerizational transformations and the influence of the R1 substituent on the process. The purpose of the study was to understand the rearrangement of these compounds and to explore the possibility of converting them into other five-membered heterocycles. The researchers concluded that the isomerization proceeds through a step involving dissociation to a quinoxalinium cation and the corresponding thioamide, and that the stability of the thiazolo[4,5-b]quinoxalines in an acidic medium is a key factor in their isomerizational transformations. The chemicals used in this process included 4-alkyl-2-phenylthiazoloquinoxalines, 2,4-dimethyl-3a,4,9,9a-tetrahydrothiazolo[4,5-b]quinoxaline, and various acids such as acetic acid, trifluoroacetic acid, and hydrochloric acid, as well as phenylthiourea for the conversion to imidazo[4,5-b]quinoxaline-2-thione.
10.1007/BF00506153
The research focuses on the synthesis and properties of substituted 10-phenyl-10-hydroxy-10H-pyrido[2,3-b]chromenes. The purpose of the study is to extend previous work on the acid-base transformations of these compounds and to determine how electronic effects are transmitted within the heterocyclic system by synthesizing derivatives with various substituents in both the benzene and pyridine rings. Key chemicals used include 2-aryloxy-3-benzoyl-6-methylpyridines (Ia-g) as starting substances, which undergo cyclization under the influence of concentrated sulfuric acid in glacial acetic acid to form the target compounds (IIa-g). The structures of these compounds were confirmed through IR and UV spectroscopy. The study concludes that the pKa+ values of the products, which range from -6.28 to -9.27, correlate with the substituent constants depending on their position, indicating the transmission of electronic effects primarily via an inductive mechanism. Additionally, the research highlights the influence of substituents on the cyclization rate and the spectral properties of the synthesized compounds.
10.1016/j.carres.2019.03.002
The study presents a metal-free, mild, and environmentally friendly method for the regioselective tandem opening of 4,6-O-benzylidene acetals to yield their corresponding 6-O-acetyl derivatives. This approach employs a 60% aqueous solution of acetic acid and demonstrates high efficiency and selectivity, applicable to both mono- and disaccharides, and suitable for large-scale synthesis. The research further applies this method to construct a building block essential for the synthesis of a trisaccharide via one-pot glycosylation reactions, showcasing its utility in carbohydrate chemistry. The study also includes detailed experimental procedures, characterization data, and a discussion on the potential applications and advantages of this green synthetic strategy, which could be beneficial for the synthesis of complex oligosaccharides and related compounds.
10.1021/jo00331a016
The study presents a chemical method for converting folic acid to pteroic acid, a valuable intermediate for synthesizing folic acid analogues and derivatives. The process involves treating folic acid with acetic anhydride to form a mixture of acetylated azlactones, which are then cleaved with mild base to yield mainly acetylated pteroic acids. Further treatment with hot base removes the acetyl groups, resulting in pteroic acid with a yield of 55-60% contaminated with folic acid. The study also discusses various side reactions and byproducts, including the formation of a pyrazine derivative from the hydrolysis of the glutamic acid moiety and the opening of the pyrimidine ring. The authors detail the experimental procedures, including HPLC analysis, UV absorption spectra, mass spectrometry, and proton NMR spectra, and provide a method for separating folic and pteroic acids using column chromatography. The research was supported by a grant from the National Cancer Institute, National Institutes of Health.


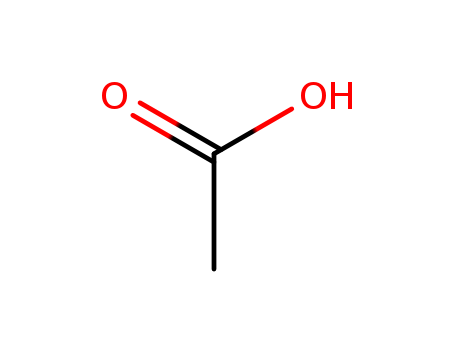
 C,
C, Xi
Xi
 C:Corrosive;
C:Corrosive;




