466
M. Prokop et al. / Electrochimica Acta 212 (2016) 465–472
coverage of Pt by these phosphorus oxoacids were examined.
According to this work, H3PO3 surface coverage of Pt increases with
rising temperature [11]. This unusual phenomenon was explained
by the tautomeric equilibria between a thermodynamically more
stable “inactive” tetrahedral and a strongly adsorbing “active”
pyramidal form of the acid [16–18].
Baudler and Schellenberg studied the electrochemical oxida-
tion of various phosphorus oxoacids as well as the H3PO4 reduction
mechanism on Pt and other metal electrodes. According to their
findings the oxidation of H3PO3 could proceed with a P(IV)
compound as an intermediate [19]. H4P2O6 is a white crystalline
compound that is stable at ambient temperature in the form of
hypophosphoric acid (H4P2O6) as a possible H3PO3 oxidation
reaction intermediate will be investigated.
2. Experimental
Extra pure chemicals, i.e. 98% phosphorous acid (Acros
Organics, extra pure), 96% sulphuric acid (Fluka, for trace analysis),
85% phosphoric acid (Acros Organics, extra pure, SLR) and 70%
perchloric acid (Acros Organics, for analysis) were used in the
experiments. Crystalline Na2H2P2O6
a method previously described by Remy and Falius [24]. The
presence of crystalline Na2H2P2O6 10H2O phase in the prepared
ꢂ10H2O was synthesised using
ꢂ
H4P2O6
ꢂ
2H2O. Hydrated hypophosphates, e.g. Na2H2P2O6
ꢂxH2O
sample was confirmed by means of X-ray diffraction using an
X’pert PRO (PANanalytical, Netherlands) diffractometer. The purity
(x = 6, 10), are also known. In aqueous solutions H4P2O6 behaves as
a weak tetrabasic acid (pKa1 = 2.2, pKa2 = 2.8, pKa3 = 7.3, pKa4 =
10.0) [20]. Therefore, hypophosphates undergo protonation to
H4P2O6 in strongly acidic aqueous solutions. The prolonged
presence of H4P2O6 in a strongly acidic environment and at
elevated temperatures leads to its disproportionation to H3PO3 and
H3PO4 according to Eq. (1). The standard redox potentials of
H4P2O6/H3PO3 and H3PO4/H4P2O6 couples are indicated in Eqs. (2)
and (3). The standard redox potential of H3PO4/H3PO3 is included
in Eq. (4) for comparison. Even at ambient temperature and when
anhydrous, H4P2O6 with a P(IV)-P(IV) bond undergoes a slow
rearrangement to form isohypophosphoric acid, containing P(III)-
O-P(V) bond, i.e. the two phosphorus atoms in different oxidation
states are connected via an oxygen atom [20]. Isohypophosphoric
acid is not stable and disproportionates to H4P2O7 and H4P2O5.
of Na2H2P2O6 10H2O was higher than 94% according to analyses
ꢂ
performed by means of thermogravimetry on STA PT 700 LT
(Linseis, Germany) with temperature scan rate of 10 ꢁC minꢀ1 from
room temperature up to 170 ꢁC, X-ray diffraction on X’pert PRO
(PANanalytical, Netherlands) and ICP-OES on Optima8000 (Perki-
nElmer, USA), while the remaining 6% probably correspond to
excess water in the salt. Before the experiment the appropriate
amount of Na2H2P2O6
10H2O was dissolved in 0.5 mol Lꢀ1 H2SO4 or
ꢂ
H3PO4 solution where Na2H2P2O6 undergoes protonation to
H4P2O6. All solutions were prepared from fresh deionised water
(conductivity 0.2 m
S cmꢀ1) made by the DIWA deionisation system
(WATEK, Czech Republic). Before each measurement the equip-
ment that would be in contact with the electrolyte and electrodes
was rinsed with distilled water and then treated with 96% H2SO4:
30% H2O2 (1:3) “piranha” solution for at least 15 hours. After
purification by “piranha” solution the equipment was thoroughly
rinsed with deionised water.
H4P2O6 + H2O ! H3PO3 + H3PO4
(1)
A pyrex glass electrochemical cell (50 cm3) tempered by a F12
cryostat (Julabo, Germany) was used for the experiments. The
measurements were performed in a three-electrode arrangement.
Pt foil (7 cm2) and Hg/Hg2SO4 in K2SO4(sat.) solution (MSE) were
used as the counter-electrode and reference electrode, respective-
ly. All potentials in this paper refer to this reference electrode. The
reference electrode was separated from the electrolyte by a double
liquid junction, the first was filled with supporting electrolyte used
in the electrochemical cell, the second with a saturated K2SO4
solution. The working electrode was planar Pt foil with a geometric
area of 1.11 cm2 and a roughness factor of about 1.25. Potentiostatic
batch electrolysis was carried out in a three-electrode arrangement
in a glass electrolytic cell with a Pt mesh (approx. 50 cm2) working
electrode and Pt wire counter-electrode separated from the
electrolyte by a ceramic frit. The same reference electrode was
used as in the voltammetry experiments. The electrolyte solution
was agitated by a PTFE stirrer (cross-like shape, 2 cm wide, rotation
speed 500 RPM). The applied potential was 0.1 V vs. MSE. Prior to
each experiment the working electrode was rinsed thoroughly
with deionised water and cycled for at least one hour between ꢀ0.7
and 0.9 V in corresponding electrolyte at a potential scan rate of
100 mV sꢀ1 After cycling, the used electrolyte was replaced by a
fresh one. All voltammetry measurements were performed with a
HEKA PG310 potentiostat. All experimental voltammograms
presented were corrected for the presence of background currents
and iR drop in the electrolyte. The electrolyte pH was measured by
an Easy Pro automatic titrator (Mettler Toledo, Switzerland) with
an EM45-BNC sensor. The concentration of H3PO3 and H3PO4 in the
electrolyte was analysed by a DIONEX ICS 1000 ion-exchange
liquid chromatograph (Thermo Scientific, USA). An IonPac AS4A-SC
4 mm analytical column with an IonPac AG4A-SC 4 mm guard
column and ASRS-ULTRA II (4 mm) anion self-regenerating
supressor were used. The mobile phase was 1.8 mmol Lꢀ1
Na2CO3 + 1.7 mmol Lꢀ1 NaHCO3 aqueous solution.
H4P2O6 + 2H+ + 2eꢀ Ð 2H3PO3 EH0 P O =H PO = ꢀ0.274 V vs. MSE (2)
4
2
6
3
3
2H3PO4 + 2H+ + 2eꢀ Ð H4P2O6 + 2H2O EH0 PO =H P O = ꢀ1.587 V vs.
3
4
4
2
6
MSE
(3)
2H3PO4 + 2H+ + 2eꢀ Ð 2H3PO3 EH0 PO =H PO = ꢀ0.930 V vs. MSE (4)
3
4
3
3
So far almost no information is available on H3PO3 oxidation
kinetics, except for the work of Trasatti and Alberti who studied the
kinetics of H3PO3 oxidation in 0.5 mol Lꢀ1 H2SO4 aqueous solution
at 25 ꢁC mainly on a bulk Pd electrode [21]. They proposed H3PO3
oxidation to a P(IV) product via catalytic dehydrogenation of H3PO3
on a Pd surface, followed by electrochemical oxidation of the
adsorbed hydrogen. The catalytic dehydrogenation to the P(IV)
compound was suggested as the rate-determining step. The P(IV)
product subsequently disproportionates to H3PO3 and H3PO4 [21].
Trassati and Alberti also briefly studied H3PO3 oxidation on a Pt
electrode, however no kinetic information was provided in their
publication [15]. They suggested that H3PO3 oxidation on a Pt
electrode proceeds by the same mechanism as on a Pd electrode.
However, this conclusion was not supported by sufficient
experimental data, and when large differences in hydrogen
adsorption energies on Pt and Pd are taken into account
(approximately ꢀ45 and ꢀ36 kJ molꢀ1 for Pd-H and Pt-H bond
respectively) their theory still remains open [22,23].
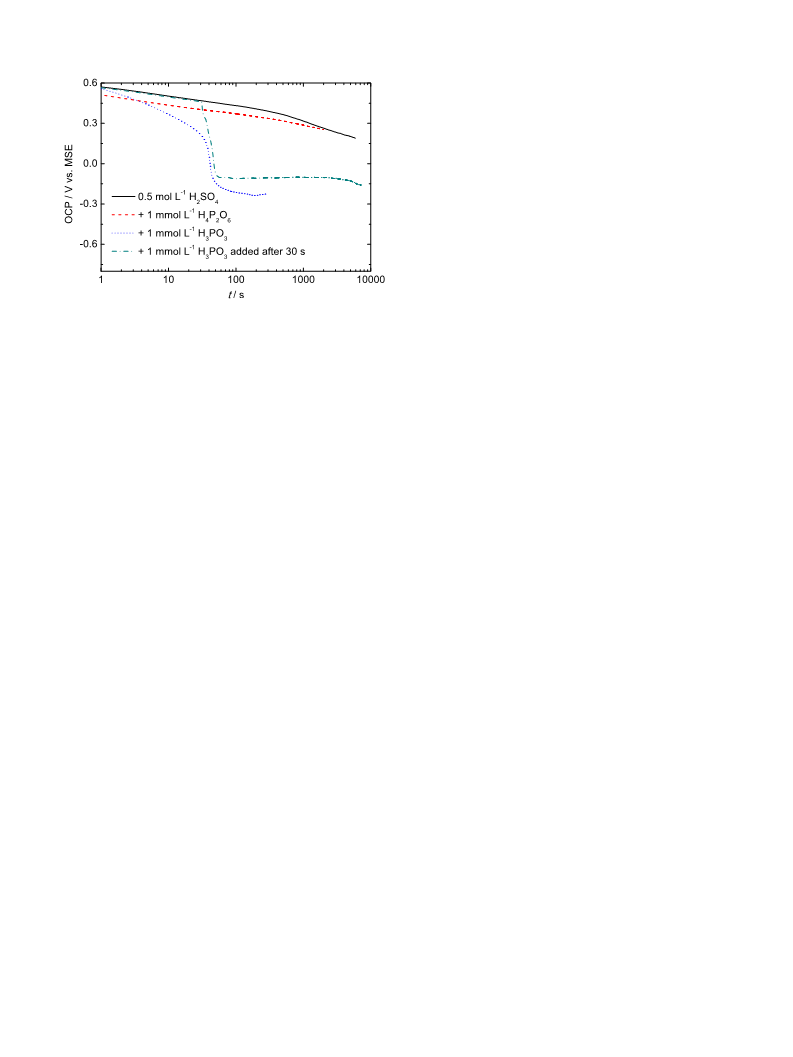
The main goal of the present study is to extend the findings
presented in our previous work [11], to include information on
H3PO3 adsorption in the presence of SO42ꢀ, HSO4ꢀ and ClO4ꢀ in the
electrolyte solution. The kinetics of H3PO3 electrochemical
oxidation will be determined for a polycrystalline Pt electrode
and for 0.5 mol Lꢀ1 H2SO4 aqueous solution at various temper-
atures. Furthermore, the electrochemical behaviour of

 Prokop
Prokop