Refernces
10.1021/jo0483466
The study investigates the competition between alkenes in intramolecular ketene-alkene [2+2] cycloaddition reactions, focusing on determining the electronic and tethering requirements for chemoselective reactions. The researchers conducted a series of experiments to understand the factors influencing the reaction rates, particularly the ring size effect and the electronic character of alkenes. They found that five-membered rings form more readily than six-membered rings and calculated the Hammett constant (F) for these cycloadditions to be -1.39, indicating that electron-donating groups accelerate the reaction. Key chemicals used in the study include ketenes, various alkenes with different electronic properties and tether lengths, and reagents like oxalyl chloride for acyl chloride formation and triethylamine for deprotonation to generate ketenes. These chemicals served to construct model compounds for the competition experiments, allowing the researchers to probe the factors affecting the chemoselectivity and kinetics of the cycloaddition reactions, which is crucial for developing new synthetic methodologies using sequential ketene cycloadditions.
10.1021/jo00021a014
The research focuses on the modification of the Bischler-Napieralski reaction for the synthesis of 3-aryl-3,4-dihydroisoquinolines. The purpose of this study was to address the inefficiencies of the traditional Bischler-Napieralski reaction in synthesizing 3-arylisoquinolines, which are important intermediates for the synthesis of various isoquinoline alkaloids and potential medicinal agents. The researchers successfully developed a method that avoids the elimination of the amide group as a nitrile via the retro-Ritter reaction by converting it to an N-acyliminium intermediate with oxalyl chloride-FeCl3. This modification resulted in the formation of 3,4-dihydroisoquinolines in moderate to high yields. The chemicals used in the process include (1,2-diphenylethyl)amides, oxalyl chloride, FeCl3, and various amide derivatives such as formamide, acetamide, benzamide, and phenylacetamide. The study concluded that this new method offers a highly effective synthetic route for the asymmetric synthesis of natural products and medicinal agents containing the 3-arylisoquinoline ring system and provides an alternative, mild method for the preparation of simple 3,4-dihydroisoquinolines.
10.1016/S0968-0896(01)00088-8
The research aimed to evaluate the stereoselectivity of chiral analogues of the endogenous cannabinoid receptor ligand, arachidonylethanolamide (anandamide), in binding to CB1 and CB2 cannabinoid receptors and their metabolic stability. The study synthesized several chiral anandamide analogues with methyl groups introduced at the 2,1 and 20 positions using asymmetric synthesis. The researchers found that the introduction of a single 2-methyl group increased affinity for CB1, but only modestly improved metabolic stability. However, the 2,1-dimethyl analogues exhibited high enantio- and diastereoselectivity, with (R)-N-(1-methyl-2-hydroxyethyl)-2-(R)-methyl-arachidonamide (4) showing the highest CB1 receptor affinity. The study concluded that a partial CB1 receptor site model could be proposed, featuring two hydrophobic pockets capable of accommodating 1- and 2-methyl groups. Key chemicals used in the process included arachidonic acid, various amino alcohols, and chiral auxiliaries like 4-benzyl-2-oxazolidinone, along with reagents such as oxalyl chloride, lithium hydroxide, and phenylmethanesulfonyl fluoride (PMSF) for enzyme inhibition during binding assays.
10.1016/j.tetlet.2004.07.085
The research focuses on the design and synthesis of novel dendritic cores based on thiacalix[4]arene derivatives, which possess potential applications in supramolecular chemistry due to their unique properties such as complexation ability towards transition metals and chemical modifiability. The purpose of the study was to overcome the steric hindrance issues encountered during the derivatization of thiacalix[4]arene derivatives to create dendritic cores with amino surface groups. The researchers successfully applied a synthetic strategy that involved the use of benzylic spacers to withdraw carboxyl groups from the thiacalix[4]arene moiety, allowing for the realization of novel thiacalix[4]arenes bearing two or four protected lysine units. Key chemicals used in the process included thiacalix[4]arene tetraacids, oxalyl chloride, mono-Boc-protected 1,2-diaminoethane, mono-Boc-protected 1,4-phenylenediamine, methyl p-bromomethylbenzoate, and CDI (1,1,3,3-tetramethyluronium chloride).
10.1016/j.tet.2007.10.025
The study explores the use of 1,3-bis(silyloxy)alk-1-enes as starting materials, which are reacted with oxalyl chloride to form the desired butenolides. The research highlights the efficiency and simplicity of this one-pot approach, which avoids the need for multiple reaction steps and purification processes. The synthesized butenolides have potential applications in pharmaceuticals and natural product chemistry due to their structural similarity to various biologically active compounds. The study also investigates the influence of different substituents on the reaction yield and the stability of the resulting compounds. Overall, the one-pot cyclization method provides a convenient and effective route for the synthesis of 4-alkoxycarbonyl-butenolides, contributing to the development of new synthetic strategies in organic chemistry.
10.2174/157340612802084270
The research focuses on the synthesis and evaluation of nine new indolyl glyoxamide derivatives (3a-i) as potential inhibitors of HIV-1. The compounds were synthesized by reacting benzylindoles (1) with oxalyl chloride to form glyoxylyl chlorides (4), which were then reacted with phenoxy phenylamines (2a or 2b) in the presence of triethylamine. The synthesized compounds were evaluated for their inhibitory activity against HIV-1 replication in acutely infected C8166 cells. The results showed that compounds 3e and 3h exhibited potent anti-HIV-1 activity with EC50 values of 6.83 and 4.35 μg/mL, and TI values of >27.15 and 49.45, respectively. The study also revealed that the introduction of substituents, particularly halogen atoms at specific positions, significantly influenced the compounds' activity. The research highlights the potential of these indolyl glyoxamide derivatives as new anti-HIV-1 agents.
10.1002/anie.201007194
The study presents a novel three-component synthesis of ynediones through a glyoxylation/Stephens–Castro coupling sequence. Copper(I) iodide (CuI) acts as the catalyst, while oxalyl chloride serves as the carbonylating agent. Terminal alkynes are the coupling partners, and electron-rich heterocycles such as indoles, pyrroles, pyrazoles, thiophenes, and furans are the nucleophilic substrates. The reaction proceeds in ethereal solvents like THF, DME, or 1,4-dioxane, and is optimized with 1.0 equivalent of oxalyl chloride, 5 mol% of CuI, 1.0 equivalent of terminal alkyne, and 3.0 equivalents of triethylamine. The method allows for the functionalization of various heterocycles in a mild, one-pot procedure, yielding densely functionalized ynediones that can serve as versatile intermediates for the synthesis of pharmaceutically relevant heterocycles. The study also demonstrates the extension of this sequence to four-component syntheses, incorporating additional nucleophiles to form more complex heterocyclic products such as enaminediones, quinoxalines, indoloyl pyrazoles, and indoloyl pyrimidines.

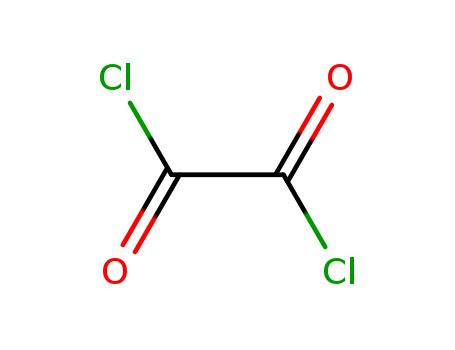

 C,
C,  T
T





