Refernces
10.1021/om100673e
The study presents the synthesis and characterization of silver and palladium complexes with xanthene-based N-heterocyclic carbene-oxazoline ligands. The researchers synthesized xanthenes with two different donor ligand units, N-heterocyclic carbene (NHC) and oxazoline, and prepared silver complexes 15 and 19 by reacting the xanthene derivatives with Ag2O. They also synthesized palladium complexes 16 and 20 by reacting the corresponding silver complexes with Pd(PhCN)2Cl2. X-ray crystallographic analysis revealed that complexes 15-PF6, 16, and 20 adopted trans configurations, while the Ag atom of 15-Cl coordinated only to the NHC ligand. The study provides insights into the structural analysis of these complexes, which could be significant for the development of catalysts for various reactions, particularly asymmetric synthesis.
10.1021/jp907080r
The research focuses on the photoactivation of azido push-pull fluorogens, which are non-fluorescent chromophores that can be converted into bright fluorescent labels suitable for single-molecule imaging. The experiments involve illuminating these aryl azide-containing fluorogens, triggering a photochemical conversion to aryl amines, which restores charge-transfer absorption and fluorescence. The study characterizes photophysical parameters such as photoconversion quantum yield, photostability, and turn-on ratio for a variety of push-pull chromophores. The research also includes the synthesis of different azido push-pull fluorogens and their photoactivation in different environments, including living cells, to demonstrate their potential for super-resolution microscopy and fluorogenic photoaffinity labeling. The analyses used include UV-vis and fluorescence spectroscopy, NMR, HPLC-MS, and microscopy techniques to confirm the conversion of non-fluorescent azido fluorogens to fluorescent amino fluorophores and to measure their photophysical properties.
10.1021/ol2009939
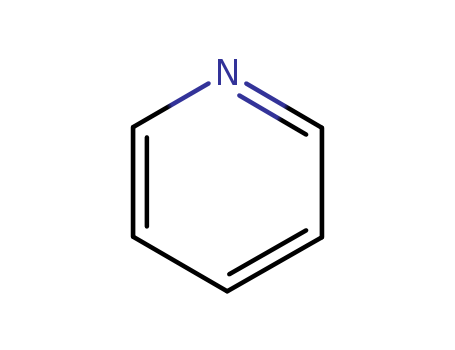
The study focuses on the development of an iron-catalyzed cycloaddition reaction between alkynenitriles and alkynes to synthesize a variety of substituted pyridines. The researchers utilized Fe(OAc)2 as a catalyst, in combination with an electron-donating, sterically hindered pyridyl bisimine ligand (L12), to facilitate this reaction. The purpose of these chemicals was to explore and establish an efficient method for the formation of pyridine derivatives, which are important in organic chemistry due to their wide range of applications. The study also involved the evaluation of various ligands to optimize the reaction conditions, ultimately achieving good yields of pyridine products under the optimized conditions. This work contributes to the expanding field of iron-based catalysis, providing a more accessible and cost-effective approach to pyridine synthesis compared to traditional methods.
10.1021/ol202691b
The study presents a novel tandem one-pot method for the synthesis of polysubstituted pyridines, which are important components in natural products, pharmaceuticals, and functional materials. The process involves the sequential reactions of nitriles with Reformatsky reagents and 1,3-enynes, utilizing the Blaise reaction intermediate. The chemicals used include various aromatic nitriles, Reformatsky reagents with different R2 groups, and a range of 1,3-enynes. These chemicals serve to construct pyridine rings with controllable substitution patterns around the pyridine core, offering a flexible and efficient synthetic approach. The purpose of these chemicals is to enable the selective control of substitution patterns in the pyridine moiety through a series of reactions that include regio- and chemoselective addition, isomerization, cyclization, and aromatization. This method not only simplifies the synthetic steps but also minimizes waste generation, and it allows for the divergent construction of two different heterocyclic rings, pyridine and pyridone, within a single molecule.
10.1246/bcsj.66.1844
The research aimed to optimize the conditions for the acylation of N-carboxy-α-dehydroamino acid anhydride (NCA) with N-protected α-amino acids (AA) to further develop the usefulness of NCA. The study examined various AA or dipeptides as a C-component in the acylation of NCA, followed by condensation with an AA methyl ester as an N-component. The researchers found that the use of dicyclohexylcarbodimide (DCC) and pyridine as a base in CH2Cl2 solvent led to the highest yield and specific rotation of the desired product, Boc-Phe-APhe-NCA (2k). The study also explored the Z-configurational structures during N-acylation and confirmed their maintenance. Key chemicals used in the process included Boc-AA-OH, DCC, pyridine, and various protected AA-OH compounds, along with DMAP as a superior catalyst to pyridine. The conclusions emphasized the successful isolation of N-protected AA-NCA in pure form and the development of new one-pot syntheses of dehydropeptides with high yields.
10.1039/c1cc16079f
The study reports on the catalytic asymmetric phospha-Michael reaction of dialkyl phosphine oxides with b,b-disubstituted a,b-unsaturated carbonyl compounds. The researchers, including Depeng Zhao and Rui Wang, aimed to construct tetrasubstituted carbon stereocenters, a challenging objective in chemical synthesis. They used dialkyl phosphine oxides as nucleophiles and various b,b-disubstituted a,b-unsaturated carbonyl compounds, including enones and N-acylpyrroles, as acceptors. The reaction was catalyzed by a zinc–bis-ProPhenol complex, with thienyl-bis-ProPhenol ligand (L2) showing superior performance. The optimized conditions involved using toluene as solvent, pyridine as an additive, and conducting the reaction at 40°C. The products were obtained in high yields (up to 98%) with excellent enantioselectivities (up to 99% ee). The study demonstrated the effectiveness of dialkyl phosphine oxides as reactive nucleophiles in this context and explored the scope of substrates, achieving consistently high enantioselectivities across a range of aromatic and aliphatic substrates.
10.1021/ic990737t
The study explores the pincer ligand chemistry of Ru(II) with a secondary amine donor, focusing on the synthesis and properties of various Ru(II) complexes coordinated with bis(tert-butylaminomethyl)pyridine (N2py). The researchers reacted N2py with RuCl2(PPh3)3 to obtain two isomers of Ru(N2py)Cl2(PPh3) (5), and with RuCl2(DMSO)4 to produce isomerically pure Ru(N2py)Cl2(DMSO) (6). The PPh3 in 5 can be replaced by CO, P(OPh)3, or pyridine, yielding Ru(N2py)Cl2(CO) (9), Ru(N2py)Cl2(P(OPh)3) (7), and Ru(N2py)Cl2(pyridine) (8), respectively. The chlorides in 9 can be replaced by CF3SO3- to form Ru(N2py)(CF3SO3)2(CO) (10). The study investigates the isomer structure preferences and the reactivity of these complexes, such as the oxidation of N2py in 8 by O2 to form a coordinated imine. The findings contribute to the understanding of the factors controlling the reactivity and stability of unsaturated Ru(II) complexes, and provide insights into the comparison of nitrogen and phosphorus donors in pincer ligand chemistry.
10.1016/S0040-4039(98)01853-X
The research explores the use of methyitrioxorhenium (CH3ReO3, also known as MTO) in combination with hydrogen peroxide (H2O2) and pyridine as a co-catalyst for the selective epoxidation of various monoterpenes. The purpose is to develop an efficient and selective method for epoxidation that can be used to produce valuable compounds for fragrances, flavors, and pharmaceuticals. The study finds that pyridine is crucial for suppressing isomerization and rearrangement of the substrates, allowing for high yields of epoxides with minimal side reactions. The reaction conditions, including solvent choice and temperature, can be adjusted to achieve either single or double epoxidation or even direct rearrangement products. The key chemicals used are MTO as the catalyst, H2O2 as the oxidant, and pyridine as the co-catalyst. The research concludes that this system is highly effective for epoxidation of terpenic substrates, offering significant advantages over traditional peracid methods in terms of product stability and yield.
10.1002/chem.202001639
The study presents an innovative approach to the enantioselective generation of quaternary carbon stereocenters through the addition of substituted cyanoacetate esters to acetylenic esters, utilizing a chiral enantiopure cobalt(III) complex as a hydrogen bond donor catalyst. The research successfully identifies the cobalt complex Δ-[Co((S,S)-dpen)3]3+ 2Cl–B(C6F5)4– (where dpen = 1,2-diphenylethylenediamine) as an effective catalyst for this reaction, achieving high enantioselectivities (up to 98% ee) and Z/E selectivities (up to 99:1) under optimal conditions. The study also explores the scope of the reaction with various substrates, revealing the catalyst's versatility and its potential for synthetic applications. Additionally, the research delves into the mechanism of the reaction, suggesting that hydrogen bonding between the catalyst's NH groups and the substrates plays a crucial role in the catalytic process. This work significantly advances the field of enantioselective catalysis, providing a new tool for the synthesis of complex molecular structures with quaternary carbon stereocenters.
10.1016/S0040-4020(00)00626-8
The study presents an eco-friendly and cost-effective method for the tosylation of alcohols and selective monotosylation of diols using p-toluenesulfonic acid with metal-exchanged montmorillonite clay as a catalyst. The Fe3+-montmorillonite clay demonstrated the highest effectiveness among the tested catalysts, outperforming Zn2+, Cu2+, Al3+-exchanged montmorillonites and K10 montmorillonite. This method allows for the regioselective tosylation of diols to monotosylated derivatives with high purity, favoring the primary hydroxy group in the presence of secondary hydroxy groups. The catalyst's reusability over several cycles was consistent, as shown in the tosylation of cyclohexanol. This approach minimizes by-product formation, typically just water, and offers advantages such as ease of catalyst recovery, recyclability, and enhanced stability compared to traditional methods using sulfonyl chloride or anhydride with organic bases.
10.1021/acs.orglett.0c02635
The study presents the development of a catalytic system for the C-alkylation of N-heterocyclic compounds, such as pyridine, pyrimidine, pyrazine, quinoline, quinoxaline, and isoquinoline, using alcohols. The process is based on a hydrogen-borrowing approach and utilizes [Cp*IrCl2]2 as the catalyst precursor, combined with potassium t-butoxide and 18-crown-6-ether. This method is environmentally friendly as it only produces water as a byproduct. The researchers optimized the reaction conditions and demonstrated the system's versatility by applying it to various substrates, achieving good to excellent yields. The study also proposed a possible reaction mechanism involving three steps: hydrogen transfer from alcohol to iridium catalyst, cross-aldol-type condensation, and transfer hydrogenation. The developed catalytic system is expected to contribute to the synthesis of pharmaceuticals and functional materials.
10.1021/ja00212a030
The study presented in the document investigates the biomimetic oxidation of 1,2-diols using molecular oxygen in the presence of iron-porphyrin catalysts, mimicking the function of metal-containing oxidases and oxygenases found in biological systems. The researchers utilized a catalytic system comprising an iron-porphyrin complex, 1-benzyl-3-carbamoyl-1,4-dihydropyridine (BNAH), and molecular oxygen to selectively cleave the carbon-carbon bonds of aryl-substituted ethane-1,2-diols at room temperature, producing aldehydes or ketones as the main oxidation products. The reaction rates were influenced by the steric hindrance of substituents in both the catalysts and diols, and no significant differences in reactivities were observed between the two stereoisomers (meso and dl) of the diols. The study provides insights into the mechanism of the diol cleavage reaction, which involves the initial binding of the diol to the active catalyst forming an intermediate complex, followed by a rate-determining breakdown step in the catalytic cycle. The findings have implications for understanding the activation of molecular oxygen and oxygen atom transfer to organic substrates, processes that are crucial for cytochrome P-450 in biological systems.
10.1002/anie.201107136
The research focuses on the Rhodium(I)-catalyzed carbon-carbon (C-C) bond cleavage in aryl ketones, directed by a pyridine group, to construct biaryls and alkenyl/alkyl arenes. The study explores an alternative synthetic method that leverages the inherent stability of carbon-carbon linkages, which is a challenging field in organic synthesis. The experiments involved the use of various catalytic systems, with a particular focus on [(CO)2Rh(acac)] as the most efficient catalyst. The reaction conditions were optimized, and a broad substrate scope was tested, including different substituents on the phenyl group and various alkyl aryl ketones. The analyses used to evaluate the outcomes included 1H NMR to determine the ratio of monoaryl/diaryl products and to confirm the absence of unwanted side products. The study demonstrated that the method is not only efficient for synthesizing biaryls but also fundamentally important for understanding the reactivity of inert C-C bonds.
10.1021/jf034067s
The study focuses on the synthesis and herbicidal activity of two series of 2-cyano-3-substituted-pyridinemethylaminoacrylates, which are designed as inhibitors of photosystem II (PSII) electron transport, targeting the disruption of photosynthetic electron transport in weeds. The chemicals used in the study include 2-cyanoacrylates with substituted pyridine rings, which were synthesized to replace phenyl groups in the parent compounds, with the aim of enhancing herbicidal activity. These compounds were confirmed through various analyses such as 1H NMR, elemental, IR, and mass spectrum analyses. The study evaluated the herbicidal activities of these compounds, finding that some exhibited excellent activity even at a low dose of 75 g/ha, and were particularly safe for use around corn, a major crop in China. The research aimed to understand the structure-activity relationship of these compounds to optimize their herbicidal properties.


 T,
T, N,
N, F,
F, Xn
Xn
 F:Flammable;
F:Flammable;




