M. Ruiz et al. / Tetrahedron 68 (2012) 705e710
709
Anal. Calcd. C14H17NO: C, 78.10; H, 7.96; N, 6.51. Found: C, 78.04; H,
7.66; N, 6.58.
and evaporated to afford the corresponding unprotected
compound.
Method C: DBU (4 equiv) and water (2 equiv) was added to a so-
lution of 1-pivaloyl derivatives (1 equiv) in THF (3 mLꢂmmol) and
was stirred to reflux until the disappearance of the starting indole.
The reaction mixture was eluyed with EtOAc (10 mL) and was
washed with a saturated aqueous NH4Cl solution (4 mLꢂmmol). The
aqueous layer was extracted with EtOAc (2ꢂ15 mL). The combined
organic layers were dried over Na2SO4 and evaporated to afford the
corresponding unprotected compound.
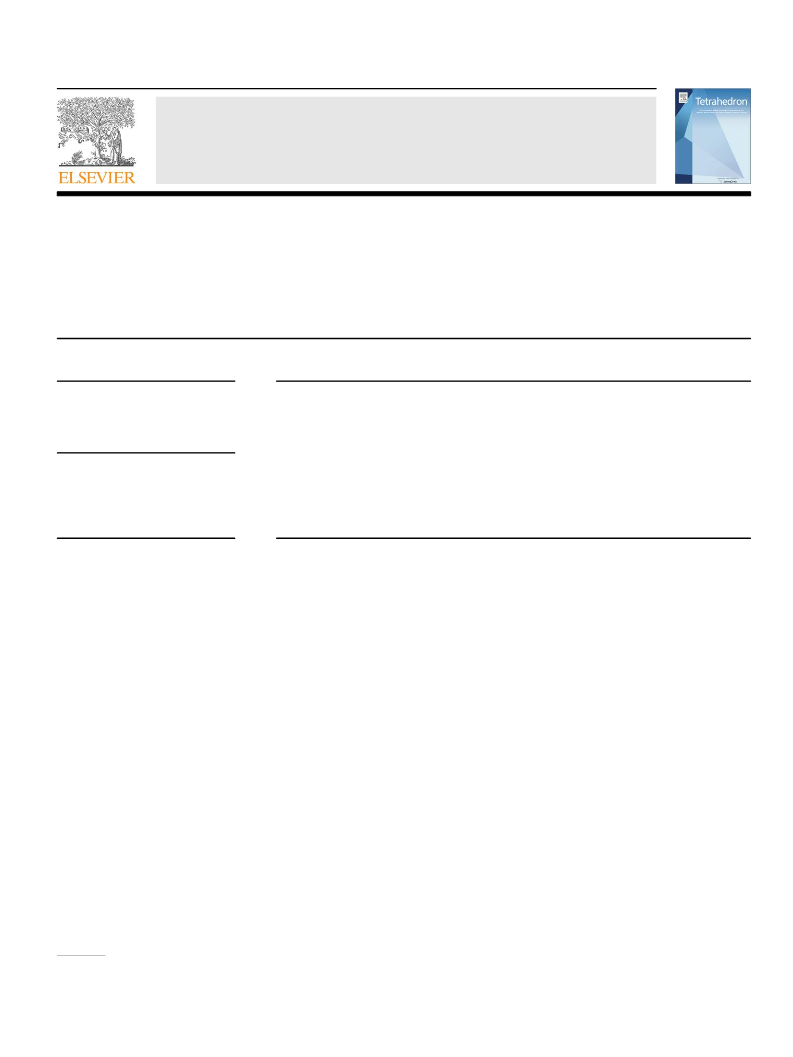
4.2.13. 4-Bromo-1-pivaloylindole (3m). Compound 3m was pre-
pared in 2.5 mmol scale using method A. Yield: 0.35 g (50%), as
a white solid. Mp 65e66 ꢁC (lit.,22 61e63 ꢁC); IR (NaCl) nmax: 1701.4
(CO); 1418 and 1305.4, 1173.6 (CeBr) cmꢀ1
;
1H NMR (CDCl3,
250 MHz)
d
: 8.50 (d, J¼8.30 Hz,1H, H-7); 7.82 (d, J¼3.8 Hz,1H, H-2);
7.75 (d, J¼7.7 Hz, 1H, H-5); 7.23 (t, J¼8 Hz, 1H, H-6); 6.73 (d,
J¼3.8 Hz, 1H, H-3); 1.56 (s, 9H, C(CH3)3); 13C NMR (CDCl3, 63 MHz)
d: 178.1 (CO); 138.1 (C-7a); 130.9 (C-3a); 127.4, 127.1 and 126.9 (C-2,
Indole derivatives thus obtained were commercial compounds,
with the exception of compounds 2,13 4j23 and 5. Characterization
data for the latter are given below.
C-6 and C-7); 117.2 (C-7); 115.3 (C-4); 108.9 (C-3); 42.3 (C(CH3)3);
29.5 (C(CH3)3). Anal. Calcd. C13H14BrNO: C, 55.73; H, 5.04; N, 5.00.
Found: C, 55.55; H, 4.96; N, 5.10.
4.3.1. 1-(1H-indol-2-yl)-3,3-dimethylbutan-2-one (5). Red solid. Mp
4.2.14. 9-Pivaloylcarbazole (6a). Compound 6a was prepared in
2.4 mmol scale using method A. Yield: 0.58 g (98%), as a white solid.
IR (NaCl) nmax: 1691 (NCO) cmꢀ1. Mp 244e245 ꢁC; 1H NMR (CDCl3,
55 ꢁC; IR (NaCl) nmax: 3367.1 (NH), 1702.6 (CO) cmꢀ1 1H NMR
;
(CDCl3, 250 MHz)
d
: 8.93(s,1H, NH); 7.59 (d, J¼7.5 Hz, 1H, H-7); 7.37
(d, J¼8.1 Hz, 1H, H-4); 7.23e7.05 (m, 2H, H-5 and H-6); 6.35 (s, 1H,
250 MHz)
d
: 7.93 (d, J¼7.5 Hz, 2H, H-4 and H-5); 7.58 (d, J¼7.5 Hz,
H-2); 4.02 (s, 2H, CH2); 1.27 (s, 9H, C(CH3)3); 13C NMR (CDCl3,
2H, H-1 and H-8); 7.35 (td, J¼7.5 and 1.3 Hz, 2H, H-2 and H-7); 7.22
63 MHz) d: 213.5 (CO); 136.8 (C); 132.4 (C); 128.5 (C); 121.9 (CH);
(td, J¼7.5 and 1.3 Hz, 2H, H-3 and H-6); 1.43 (s, 9H, C(CH3)3); 13C
120.3 (CH); 120.1 (CH); 11.3 (CH); 101.6 (CH); 45.4 (C(CH3)3); 35.9
(CH2); 26.5 (C(CH3)3). Anal. Calcd for: C14H17NO: C, 78.10; H, 7.96; N,
6.51. Found: C, 78.34; H, 7.80; N, 6.51.
NMR (CDCl3, 63 MHz) d: 184.0 (CO); 139.1 (C-8a and C-8b); 126.3 (C-
2 and C-7) 124.6 (C-4a and C-4b); 121.8 (C-3 and C-6); 120.0 (C-4 and
C-5); 113.7 (C-1 and C-8); 43.6 (C(CH3)3); 28.3 (C(CH3)3). Anal. Calcd.
C17H17NO: C, 81.24; H, 6.92; N, 5.57. Found. C, 77.06; H, 7.40; N, 8.69.
Acknowledgements
4.2.15. 3-Methyl-9-pivaloyl-1,2,3,4-tetrahydrocarbazole
(6b). Compound 6b was prepared in 3.7 mmol scale using method
A. Yield: 0.86 g (85%), as an oil. IR (NaCl) nmax: 1701 (CO) cmꢀ1; 1H
We thank MEC (grant CTQ2009-12320-BQU) and UCM (Grupos
ꢀ
de Investigacion Consolidados, grant GR35/10-A-920234) for fi-
nancial support. A predoctoral fellowship from UCM to M.R. is also
gratefully acknowledged.
NMR (CDCl3, 250 MHz) d: 7.47e7.40 (m, 2H, H-5 and H-8); 7.21 (td,
J¼7.2 and 2.1 Hz, 1H, H-6); 7.17 (td, J¼7.2 and 1.6 Hz, 1H, H-7);
2.89e2.66 (m, 3H, H-4, 2ꢂH-1); 2.35e2.23 (m, 1H, H-4); 2.03e1.96
(m, 2H, H-2 and H-3); 1.65e1.50 (m, 1H, H-2); 1.48 (s, 9H, C(CH3)3);
Supplementary data
1.18 (d, J¼6.5 Hz, 3H, CH3); 13C NMR (CDCl3, 63 MHz)
d: 184.7 (CO);
Supplementary data associated with this article can be found in
clude MOL files and InChiKeys of the most important compounds
described in this article.
135.7 (C-8a); 135.6 (C-9a); 128.8 (C-4b); 122.0 (C-6); 120.9 (C-7);
118.0 (C-5); 114.4 (C-4a); 112.8 (C-8); 43.4 (C(CH3)3); 31.5 (C-2); 29.3
(C-4); 28.9 (C-3); 28.3 (C(CH3)3); 23.8 (C-1); 21.6 (CH3). Anal. Calcd.
C18H23NO: C, 80.26; H, 8.61; N, 5.20. Found: C, 79.99; H, 8.33; N 5.16.
References and notes
4.3. General procedures for the deprotection of N-
pivaloylindoles
1. For reviews of the applications of the privileged structure concept in drug
discovery, see: (a) DeSimone, R. W.; Currie, K. S.; Mitchell, S. A.; Darrow, J. W.;
Pippin, D. A. Comb. Chem. High Throughput Screen. 2004, 7, 473; (b) Duarte, C. D.;
Barreiro, E. J.; Fraga, C. A. Mini Rev. Med. Chem. 2007, 7, 1108; (c) Welsch, M. E.;
Snyder, S. A.; Stockwell, B. R. Curr. Opin. Chem. Biol. 2010, 14, 1.
2. For the first mention of the privileged structure concept, see: Evans, B. E.; Rittle,
K. E.; Bock, M. G.; DiPardo, R. M.; Freidinger, R. M.; Whitter, W. L.; Lundell, G. F.;
Veber, D. F.; Anderson, P. S.; Chang, R. S. L. J. Med. Chem. 1988, 31, 2235.
3. (a) Kocienski, P. J. Protecting Groups, 3rd ed.; Georg Thieme: Stuttgart, 2004; (b)
Greene, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis, 4th ed.;
Wiley-Interscience: Hoboken, New Jersey, 2007, pp 872d888.
Method A: To 1.6 M solution of butyllithium in hexanes (2 equiv)
was added dropwise
a stirred solution of diisopropylamine
(2 equiv) in dry THF (2 mLꢂmmol) at 0 ꢁC, under an argon atmo-
sphere. Stirring was continued for 10 min at 0 ꢁC, and the solution
of LDA thus prepared was added via cannula to a stirred solution of
the suitable 1-pivaloyl derivative (1 equiv) in THF (2 mLꢂmmol),
under an argon atmosphere at ꢀ78 ꢁC. When addition was com-
plete, the reaction mixture was heated in an oil bath at 40e45 ꢁC for
2 h, cooled and poured onto a saturated aqueous NH4Cl solution
(30 mL), which was then extracted with CH2Cl2 (3ꢂ15 mL). The
combined organic layers were dried over Na2SO4 and evaporated,
and the residue was chromatographed on silica gel, eluting with 9:1
EtOAcepetroleum ether. Evaporation of the mobile phase yielded
the deprotected indole, which was identical in all respects to the
commercially available sample employed as starting material for
the protection reaction.
4. For representative examples, see: (a) He, F.; Snider, B. B. Synlett 1997, 483; (b)
Yokoyama, F.; Sugiyama, H.; Aoyama, T.; Shiori, T. Synthesis 2004, 1476.
5. Baran, P. S.; Guerrero, C. A.; Corey, E. J. Org. Lett. 2003, 5, 1999.
6. Teranishi, K.; Hayashi, S.; Nakatsuka, S.-I.; Goto, T. Synthesis 1995, 506.
7. For a recent example (although without final deprotection), see: Stuart, D. R.;
Villemure, E.; Fagnou, K. J. Am. Chem. Soc. 2007, 129, 12072.
ꢀ
ꢀ
ꢀ
8. (a)Moldvai,I.;Temesvari-Major, E.;Balazs, M.;Gacs-Baitz, E.;Egyed,O.;Szantay, C.
J. Chem. Res. (S) 1999, 687; (b) Meng, C. Q.; Ni, L.; Worsencroft, K. J.; Ye, Z.; Wein-
garten, M. D.; Simpson, J. E.; Skudlarek, J. W.; Marino, E. M.; Suen, K.-L.; Kunsch, C.;
Souder, A.; Howard, R. B.; Sundell, C. L.; Wasserman, M. A.; Sikorski, J. A. J. Med.
Chem. 2007, 50, 1304; (c) For few examples of the deprotection of 2,2-dieth-
ylbutanoyl)indoles, see: Fukuda, T.; Maeda, R.;Iwao, M. Tetrahedron 1999, 55, 9151.
9. Horwell, D. C.; McKiernan, M. J.; Osborne, S. Tetrahedron Lett. 1998, 39, 8729.
10. Lee, M.; Ikeda, I.; Kawabe, T.; Mori, S.; Kanematsu, K. J. Org. Chem. 1996, 61, 3406.
11. Fujishita, T.; Yoshinaga, T. U.S. Patent 6,333,323, 2001.
Method B: A mixture of DBU (4 equiv) and water (2 equiv) was
added to a solution of the suitable 1-pivaloyl derivatives (1 equiv)
in THF (3 mLꢂmmol) and was stirred at rt until the disappearance
of the starting indole. The reaction mixture was eluyed with EtOAc
(10 mL) and was washed with a saturated aqueous NH4Cl solution
(4 mLꢂmmol). The aqueous layer was extracted with EtOAc
(2ꢂ15 mL). The combined organic layers were dried over Na2SO4
ꢀ
ꢀ
12. Moldvai, I.; Temesvari-Major, E.; Incze, M.; Szentirmay, E.; Gacs-Baitz, E.;
ꢀ
Szantay, C. J. Org. Chem. 2004, 69, 5993.
13. Teranishi, K.; Nakatsuka, S.; Goto, T. Synthesis 1994, 1018.
14. MacCoss, R. N.; Henry, D. J.; Brain, C. T.; Ley, S. V. Synlett 2004, 675.
ꢀ
ꢀ
15. Ruiz, M.; Lopez-Alvarado, P.; Menendez, J. C. Org. Biomol. Chem. 2010, 8, 4521.
16. For reviews of the chemistry of N-methylwelwistatin and related alkaloids
~
ꢀ
(welwitindolinones), see: (a) Avendano, C.; Menendez, J. C. Curr. Org. Synth.

 Ruiz, Míriam
Ruiz, Míriam