E. Grobbelaar et al. / Inorganica Chimica Acta 359 (2006) 3800–3806
3805
CH3I (0.015 M) and followed at 425.0 nm. A pseudo-first-
order rate constant of 1.35 · 10ꢀ3 sꢀ1 (=k3[CH3I]) was
obtained. This rate is a factor 3 faster than the rate
obtained for k2 (4.56(5) · 10ꢀ4 sꢀ1) implying that the
pathway. This non-linear orientation was due to steric
crowding of the cod ligand‘s methylene protons above
and below the plane formed by the iridium and the ancil-
lary bidentate ligand atoms. We envisaged the same phe-
nomenon in the present complex in that the rate of initial
nucleophilic attack of the metal upon the methyl carbon
in CH3I is slowed down due to the above-mentioned steric
crowding. This may also be the reason why the solvolysis
reaction becomes more pronounced relative to the oxida-
tive addition rate.
assumption of k3[CH3I] ꢂ k made in the deduction of
ꢀ2
Eq. (4) was justified. The fact that k3[CH3I] P k2 further
strengthens the argument for the existence of the k3
pathway.
This solvent-assisted oxidative addition reaction is fur-
thermore a factor 10 slower than the comparable rate for
the direct pathway under the same conditions. Similar
results were also obtained when this experiment was
repeated with chloroform as solvent. In this case oxidative
addition via the solvent-assisted pathway was a factor 40
slower than that for the direct pathway. Oxidative addition
therefore occurs mainly via the direct pathway especially
since the solvolysis rate constants for acetone and chloro-
form (k2 = 4.56(5) · 10ꢀ4 and 8.4(1) · 10ꢀ4 sꢀ1, respec-
tively) are relatively slow.
¼
¼
The activation parameters, DH and DS , for the oxida-
tive addition of CH3I to (Bu4N)[Ir2(l-Dcbp)(cod)2] (k1
pathway) are 47(2) kJ molꢀ1 and ꢀ110(7) JKꢀ1 molꢀ1
,
respectively. The activation parameters for the reductive
elimination reaction (k pathway) were not determined
ꢀ1
because the intercept values in Table 2 are a combination
of the k and the k2/k3 pathways. The fairly negative
ꢀ1
entropy of activation together with the positive enthalpy
of activation is indicative of an associative process which
is more enthalpy than entropy driven for the k1 pathway.
The enthalpy of activation for the k1 pathway is similar
to the values which was reported for the corresponding
[Ir2(l-pz)2(cod)2] complex [23]. There are however a big dif-
ference in the entropy of activation being zero within
experimental error in the case of the [Ir2(l-pz)2(cod)2] com-
plex (15(25) J Kꢀ1 molꢀ1). Both the entropy and the
enthalpy values of activation for the (Bu4N)[Ir2(l-
Dcbp)(cod)2] complex is also in agreement with the general
activation parameters which was reported for the corre-
sponding monomeric iridium(I) complexes [26].
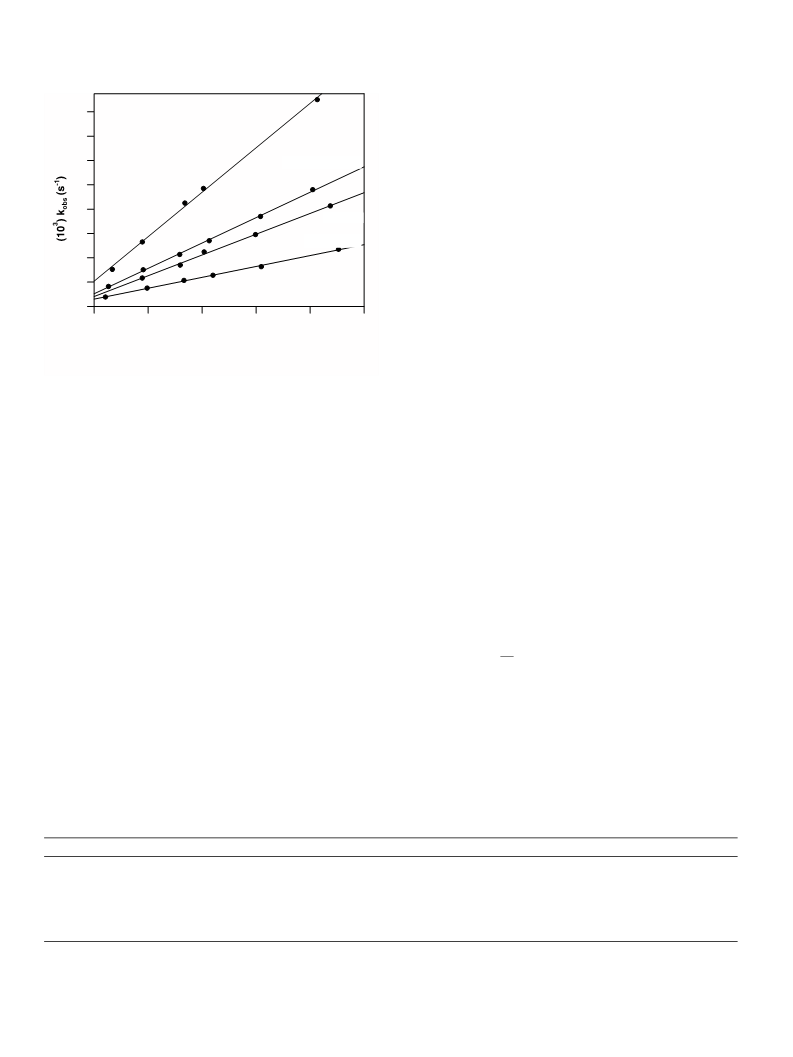
The intercept, which was obtained from the plots of the
pseudo-first-order rate constants, kobs, versus [CH3I] in
the different solvents (Fig. 3), is therefore a combination of
the solvolysis reaction (k2 pathway) and reductive elimina-
tion (k pathway). The results in Table 2 of the solvent
ꢀ1
effects show that the rate of oxidative addition depends
to a slight degree on the characteristics of the different sol-
vents. The most important being a decrease in rate con-
stants with a simultaneous increase in dielectric constant.
The observed solvent effect could however not be attrib-
uted to the donosity of the solvents.
This is in contrast to the expected solvent effect for oxi-
dative addition reactions. Normally oxidative additions
are characterised by a 20–60-fold increase in the oxidative
addition rate when the solvents are varied [21]. An example
of such an increase in the oxidative addition rate is the 20-
fold increase in the rate constants for the oxidative addition
of CH3I to [Rh(acac)(CO)(PPh3)] in different solvents [22].
A comparison of the solvent effect for the k1 pathway in this
case, with that observed for the oxidative addition of CH3I
to the corresponding [Ir2(l-pz)2(cod)2] complex shows that
the solvent effect in our case is remarkably smaller than the
reported effect [23]. The increase in the oxidative addition
rate in this study is 4-fold while in the case of [Ir2(l-pz)2-
(cod)2] there is a 16-fold increase in the rate constants.
Our results also compare with an increase of approximately
a factor 9 which was observed for the oxidative addition
reaction between the monomeric [Ir(hpt)(cod)] complex
and iodomethane and an increase of a factor 4 for the cor-
responding reaction between [Ir(AnMetha)(cod)] and
iodomethane [18].
Acknowledgements
The authors thank the South African Foundation for
Research Development as well as the Research Fund of
this University for financial support.
References
[1] A.P. Sadimenko, S.S. Basson, Coord. Chem. Rev. 147 (1996) 247.
´
[2] M.P. Garcıa, M.A. Esteruelas, M. Martin, L.A. Oro, J. Organomet.
Chem. 467 (1994) 151.
[3] M. A Esteruelas, M.P. Garcıa, A.M. Lopez, L.A. Oro, Organomet-
allics 10 (1991) 127.
[4] M.P. Garcia, A.M. Lopez, M.A. Esteruelas, F.J. Lahoz, L.A. Oro, J.
Chem. Soc., Chem. Commun. (1988) 793.
[5] M.A. Esteruelas, M. P Garcia, A.M. Lopez, L.A. Oro, Organomet-
´
´
´
´
allics 11 (1992) 702.
[6] K.A. Beveridge, G.W. Bushnell, K.R. Dixon, D.T. Eadie, S.R.
Stobart, J. Am. Chem. Soc. 104 (1982) 920.
[7] A.W. Coleman, D.T. Eadie, S.R. Stobart, J. Am. Chem. Soc. 104
(1982) 922.
The solvent effect for the oxidative addition reactions
observed here is also strongly reminiscent to that of
[Ir(hpt)(cod)] and [Ir(AnMetha)(cod)] [18] where negative
intrinsic volumes of activation, supported by a significant
reduced angle (156ꢁ) for the trans-orientated H3C–Ir(III)-
I moiety in the oxidative addition products [24,25], pointed
[8] J.L. Atwood, K.A. Beveridge, G.W. Bushnell, K.R. Dixon, D.T.
Eadie, S.R. Stobart, M.J. Zaworotko, Inorg. Chem. 23 (1984)
4050.
[9] K.A. Beveridge, G.W. Bushnell, S.R. Stobart, Organometallics 2
(1983) 1447.
[10] D.O.K. Fjeldsted, S.R. Stobart, M.J. Zaworotko, J. Am. Chem. Soc.
107 (1985) 825.
[11] J.A. Bailey, S.L. Grundy, S.R. Stobart, Organometallics 9 (1990) 536.
¼
to a linear SN2 transition state of [Irꢅ ꢅ ꢅCH3–I] for the k1

 Grobbelaar, Ebeth
Grobbelaar, Ebeth