Refernces
10.1016/j.tet.2009.01.058
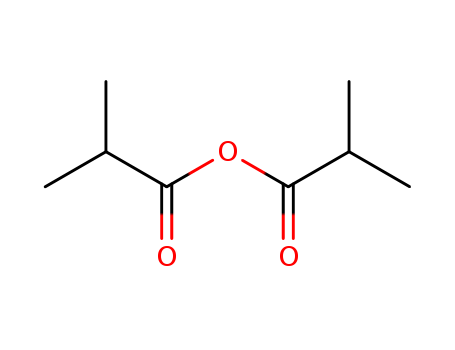
The study focuses on the nonenzymatic kinetic resolution of racemic 2,2,2-trifluoro-1-aryl ethanols, utilizing (R)-benzotetramisole as a catalyst. The aim was to achieve enantioselective acylation, which is crucial for obtaining chiral 1-substituted 2,2,2-trifluoro-ethanols, important intermediates in the synthesis of biologically active molecules. Various aryl-substituted ethanols were tested to evaluate the system's ability to differentiate between enantiomers, with a focus on the impact of different aryl groups on the enantioselectivity, as indicated by the s value. The study also optimized reaction conditions, including the choice of catalyst, acylating reagent, solvent, and reaction temperature, to maximize enantioselectivity and reaction efficiency. The chemicals used served specific purposes: (R)-benzotetramisole as the catalyst to facilitate the reaction, isobutyric anhydride as the acylating reagent to promote acylation, and diisopropyl ether as the solvent providing the appropriate polarity for the reaction. The study demonstrated that certain aryl groups, particularly phenyl and naphthyl groups, could yield high s values, indicating effective kinetic resolution. The research also provided preparative kinetic resolution examples to showcase the method's applicability in preparing enantiomerically pure 2,2,2-trifluoro-1-aryl ethanol derivatives.
10.1021/ja00204a015
The research focuses on the total synthesis of d,l-methynolide, the aglycon of the macrolide antibiotic methymycin, utilizing sulfur-mediated ring expansion technology. The purpose of this study was to develop a synthetic approach that relies on sequential 2,3-sigmatropic rearrangements of stabilized sulfonium ylides to build cyclic sulfides of varying ring sizes and to achieve remote stereocontrol through the predictable conformational properties of medium-sized ring intermediates and the stereoelectronic effect of sulfur α to ketone carbonyl. The successful route involved the ring expansion of sulfonium ylide to an eight-membered sulfide, followed by conversion to alcohol and reduction of the double bond to form the saturated analogue. Key chemicals used in the process included sulfonium ylides, sulfides, alcohols, and various reagents for protection, oxidation, and reduction steps, such as lithium ethyl boranolate (LiEt3BH), methyltriphenylphosphonium bromide, potassium tert-butoxide, and p-toluenesulfonylhydrazide, among others. The conclusions of the research detailed the successful synthesis of d,l-methynolide and a similar route to ClO-epi-methynolide, demonstrating the efficacy of the sulfur-based strategy for remote stereocontrol in complex macrocycle synthesis.
10.1248/cpb.35.2196
Yuji Oikawa, Tatsuyoshi Tanaka, Tatsuo Hamada, and Osamu Yonemitsu detail a highly stereoselective synthesis of the seco-acid (3) of the aglycone methoxylactone (1) of the 12-membered macrolide antibiotic methicillin. The synthesis started from D-glucose and involved the Wittig-Horner coupling of two fragments (i and ii). Fragment i (4) was synthesized by kinetic acetalization with p-methoxybenzylide (MP) acetal protection, while fragment ii (5) was obtained from the Prelog-Djerassi lactone-type intermediate (2) through a series of reactions including protection, hydrolysis, reduction, and oxidation. Diethyl methylphosphonate, a widely used organic synthesis reagent, was used to synthesize fragment ii (5) to introduce the phosphonate group. The coupling of these fragments formed the ring-opened acid (3), which was confirmed by nuclear magnetic resonance (NMR) and mass spectrometry. This work represents a major advance in the synthesis of complex macrolide antibiotics, demonstrating the utility of noncyclic stereocontrolled approaches and the importance of careful choice of protecting groups to achieve high stereoselectivity.
10.1039/b419335k
The research details the development of a new class of chiral 4-(pyrrolidino)-pyridine catalysts derived from (S)-proline for the kinetic resolution of sec-alcohols. These catalysts, including compounds 4 and 5, leverage both van der Waals (π) and H-bonding interactions to achieve enantioselective acylation. The study involved synthesizing these catalysts from simple starting materials like 3-carboxy-4-chloropyridine and various amines. The catalysts were evaluated in the kinetic resolution of mono-protected diols in the presence of isobutyric anhydride, with the (S)-prolinol-derived catalysts showing significant enantioselectivity. The hydroxyl group in these catalysts was found to play a crucial role in determining the selectivity of the acylation reactions. The researchers also explored the influence of different substituents and the impact of H-bonding on the selectivity, using NMR spectroscopy to investigate possible aryl-pyridinium ion π-stacking interactions. The findings suggest that these catalysts represent a novel approach to achieving remote stereochemical control in acylation reactions, with potential applications in enantioselective acyl-transfer processes.


 C
C





