Refernces
10.1021/ja00207a029
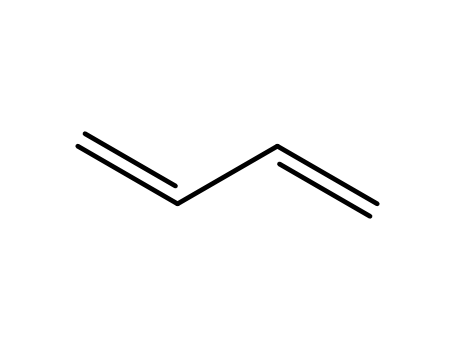
The purpose of the first part of the research was to investigate the modification of phosphorothioate in single-stranded and self-complementary oligonucleotides, with the aim of improving their efficiency in various studies, including protein binding, resonance energy transfer, structural analyses, and nucleic acid dynamics. The researchers observed that the hydrolytic stability of these triesters was influenced by pH and the length of the oligonucleotide fragments. In the second part, the researchers explored the use of cyclopropyl Grignard reagents in the synthesis of butadienes, a method that had not been previously reported. They found that this approach could yield butadienes in a simple and efficient manner, with the potential for further application in synthesis.
10.1002/anie.201915075
The study presents a novel method for synthesizing alternating copolymers composed exclusively of acrylamide units. The key to this synthesis is a specially designed divinyl monomer that includes acrylate and acrylamide moieties connected by two activated ester bonds. This design enables a "selective" cyclopolymerization process, where intramolecular and intermolecular propagation occur alternately under dilute conditions. The resulting cyclopolymer can then be transformed into alternating copolymers by reacting with various amines, yielding different acrylamide units in an alternating sequence. The study demonstrates the synthesis of ten types of alternating copolymers, some of which exhibit unique properties in solution and bulk, distinct from random copolymers. These properties are attributed to the alternating sequence of the copolymers. The chemicals involved include the divinyl monomer with activated ester bonds, amines for the cleavage reactions, and various acrylamide units that form the final copolymers. The study highlights the potential for creating sequence-controlled synthetic polymers with properties similar to natural polymers.
10.1039/c39760000371
The study investigates the flash thermolysis of trimethylsilyl 2-oxobicyclo[3.3.1]nonane-1-carboxylate (1), which leads to the formation of enol ethers (3) and (5). The primary focus is on the transient existence of a strained bridgehead enol ether (2) as an intermediate. The ester (1) is prepared by reacting the corresponding acid with trimethylsilyl chloride in the presence of pyridine. Thermolysis is conducted at various temperatures, and products are isolated based on their volatility using the 'sandwich technique' for slightly volatile compounds and direct distillation for volatile ones. The main products identified are enol ethers (3) and (5), along with their hydrolysis products, ketones (4) and (6). The study also observes the formation of butadiene and enol ether (7) at higher temperatures, suggesting they are secondary products derived from compound (5). The structures of the products are confirmed through spectroscopic data and chemical analysis. The presence of enol ethers (3) and (5) supports the formation of the strained bridgehead enol ether (2), which can undergo a [1,3] hydrogen shift to form (3) or a retro-Diels-Alder reaction to form (5).
10.1038/nchem.1882
Ryo Nakano, Shingo Ito, and Kyoko Nozaki present a novel strategy for copolymerizing carbon dioxide (CO2) and butadiene, overcoming the thermodynamic and kinetic barriers typically associated with such reactions. The authors utilize a metastable lactone intermediate, 3-ethylidene-6-vinyltetrahydro-2H-pyran-2-one, formed through a palladium-catalyzed condensation of CO2 and butadiene. This intermediate is then polymerized via free-radical polymerization to produce high molecular weight polymers with a CO2 content of 33 mol% (29 wt%). The method was successfully applied to one-pot copolymerizations and terpolymerizations, yielding polymers with potential applications as engineering plastics. The study not only provides a sustainable route for polymer synthesis but also opens up new possibilities for utilizing CO2 in the production of synthetic polymers.
10.1016/S0040-4020(98)00193-8
The research focuses on the synthesis of 11-methoxycarbonyl-13-phenyl-17-vinylgona-1,3,5(10)-trienes, which are significant compounds in the field of steroid chemistry. The study aims to develop a novel and efficient strategy for the synthesis of these complex molecules, leveraging the titanium tetrachloride-mediated dialkylation of methyl 4-oxo-4-(p-bromophenyl)butanoate ethylene ketal by 1,8-bis(trimethylsilyl)-2,6-octadiene (BISTRO). The process involves several steps, including methoxycarbonylation, alkylation by iodobenzocyclobutene, and pyrolysis, to yield the desired steroid compounds. The research concludes that this methodology offers a short and efficient route to 13-arylgonatrienes from 1,3-butadiene and benzocyclobutenol, with the added advantage of being able to modify the substituents on the phenyl group and transform the vinyl group through Wacker-type oxidation, thus enhancing the synthetic versatility of the method. Key chemicals used in this process include titanium tetrachloride, 1,8-bis(trimethylsilyl)-2,6-octadiene (BISTRO), methyl 4-oxo-4-(p-bromophenyl)butanoate ethylene ketal, iodobenzocyclobutene, and dimethyl carbonate, among others.
10.1021/jo00375a002
The research focuses on the development and utilization of recoverable, recyclable nickel(0) diene cyclooligomerization catalysts using functionalized ethylene oligomers as ligands. The purpose of this study was to create a catalyst system that maintains high product selectivity and reactivity, similar to conventional homogeneous catalysts, while also being recoverable and recyclable. The researchers synthesized various polyethylene-bound phosphite ligands and evaluated their effectiveness in cyclooligomerization reactions involving butadiene. The results demonstrated that these macromolecular ligands could mimic the chemistry of conventional ligands, achieving high catalyst activity and selectivity, with the added benefit of facilitating catalyst recovery and recycling. The chemicals used in the process included a range of phosphite ligands, nickel(II) acetylacetonate as the nickel source, and butadiene as the substrate, along with aluminum ethyl as a reducing agent. The study concluded that the product selectivity could be varied by adjusting the ligand-to-metal ratio and that the catalysts could be recycled multiple times with minimal loss of activity, highlighting the potential for these catalysts in industrial applications where catalyst recovery and recycling are economically and environmentally beneficial.
10.1016/S0022-328X(00)98506-1
The research investigates the photochemical reactions of RMn(CO)5 complexes (where R = H, C6H5, CH2C6H5) with 1,3-butadiene and 1,3-pentadiene. The study explores how these dienes insert into the Mn-C bonds, forming various Mn(CO)3(η2-enyl) complexes. Key chemicals involved include HMn(CO)5, C6H5Mn(CO)5, and C6H5CH2Mn(CO)5 as starting materials, along with 1,3-butadiene and 1,3-pentadiene as diene reactants. The reactions are induced by irradiation at 253 K, and the resulting complexes are characterized using IR and NMR spectroscopy, as well as elemental analysis. The study also examines the stabilization of enyl ligands through substitution with donor ligands like P(CH3)3 and As(CH3)3, which allows for the isolation and characterization of different isomers of the Mn(CO)3(η2-enyl) complexes. The research provides insights into the stereochemistry and configurations of these complexes, highlighting the role of different substituents and donor ligands in influencing the stability and structure of the enyl ligands.


 F+,
F+, T,
T, F,
F, N
N





