Refernces
10.1016/j.bmcl.2004.02.092
The study focuses on the design, synthesis, and evaluation of a novel class of pyrazolo[3,4-d]pyrimidines as potent inhibitors of enteroviruses, specifically coxsackieviruses. The researchers synthesized a series of these compounds and tested their antiviral activity using a plaque reduction assay. They discovered that these compounds showed remarkable specificity for human enteroviruses, with some derivatives highly effective at nanomolar concentrations. Structure-activity relationship (SAR) studies indicated that the phenyl group at the N-1 position and the hydrophobic diarylmethyl group at the piperazine significantly influenced the in vitro antienteroviral activity. Notably, compounds with a thiophene substituent, such as 20–24, exhibited high activity against coxsackievirus B3 and moderate activity against enterovirus 71, without apparent cytotoxic effects on RD cell lines. The findings highlight the potential of these compounds as new antiviral agents against enteroviral infections, for which effective treatments are currently lacking.
10.1016/j.bmc.2019.05.026
The research focuses on the synthesis and evaluation of new pyrazolopyrimidine derivatives as potential inhibitors of Leishmania amazonensis arginase, an enzyme crucial for polyamine biosynthesis in the parasite. Six derivatives with varying substituents at the 4-position of the phenyl group were synthesized and tested for their inhibitory activity against recombinant L. amazonensis arginase (LaARG). The synthesis involved reactions of phenylhydrazine with malononitrile, formic acid, and phosphorous oxychloride to obtain the desired compounds, which were then confirmed through techniques like NMR, IR, EI-MS, and HRMS. The biological evaluation included determining the IC50 values, kinetic analysis of enzyme inhibition, and molecular docking studies to understand the interaction of these compounds with LaARG. Additionally, the compounds were assessed for their cytotoxicity on mammalian macrophages and their anti-leishmanicidal activity against L. amazonensis amastigotes. The study utilized various analytical methods such as high-performance liquid chromatography (HPLC) for purity assessment and molecular modeling for structural analysis of the enzyme-inhibitor complexes.
10.1021/ol026383i
The research focuses on the synthesis of dimethylaminomethylene vinamidinium salts, which are significant synthetic intermediates, through a [2 + 2] cycloaddition route starting from γ-fluoroalkanoic acids. The primary reactants include trifluoropropanoic acid, phosphorus oxychloride (POCl3), and N,N-dimethylformamide (DMF). The reaction pathway involves the formation of an intermediate trifluoromethyl enamine, which can either react with additional POCl3 to form a vinamidinium salt or undergo a thermally driven loss of fluoride to generate an iminium ion, leading to the target product. Spectroscopic studies, including in situ IR and Raman spectroscopy, as well as 31P and 19F NMR spectroscopy, were employed to define the reaction pathway and to construct a mechanistic framework involving two cycloaddition processes. These analyses helped in understanding the reaction mechanism and the factors responsible for the formal loss of the trifluoromethyl group, ultimately providing direct access to these important vinamidiniums and identifying an unusual reactivity mode for γ-fluoroenamines.
10.1016/j.carres.2007.03.029
The research focuses on the synthesis and evaluation of fluorescently labeled and internally quenched UDP-Gal probes, which are sugar nucleotide analogs with a fluorescence emitter and a quencher. These probes were designed to assess their recognition and usage by various galactosyltransferases, enzymes crucial for the biosynthesis of mammalian oligosaccharides. The study involved detailed chemical syntheses of the UDP-Gal analogs, utilizing a range of reactants such as uridine, galactose, and phosphorus oxychloride, among others, and employing techniques like palladium-catalyzed coupling, hydrogenation, and ion-exchange chromatography. The experiments aimed to determine the rate of galactose transfer by several galactosyltransferases, including blood group B a-(1!3) galactosyltransferase, a-(1!3) galactosyltransferase, and milk bovine b-(1!4) galactosyltransferase, using the synthesized UDP-Gal analogs as substrates. Analytical methods employed to monitor the reactions and characterize the products included TLC, MALDI TOF mass spectrometry, and NMR spectroscopy. The results demonstrated that the modified UDP-Gal analogs were recognized as weak substrates by the tested galactosyltransferases, with the ability to transfer their galactose unit to the acceptor molecules.
10.1021/jm060213k
The research focuses on the synthesis and evaluation of dopamine receptor antagonists based on the benzindoloazecine structure. The study aimed to modulate the affinities for dopamine D1-D5 receptors by homologizing a lead compound, LE 300. Two homologue antagonists were synthesized, and their affinities and inhibitory activities at D1-D5 receptors were measured using radioligand binding experiments and a functional Ca2+ assay. The phenylpropyl homologue 3 showed superior selectivity and affinity for the D5 subtype with a Ki of 0.6 nM, while the indolylpropyl homologue 2 exhibited decreased affinity for all subtypes. The experiments involved the synthesis of compounds 2 and 3 through a series of chemical reactions, including the use of tryptamine, lactone 15, and various reagents such as PCl5, POCl3, and NaBH4. The synthesized compounds were then tested for their binding affinities and inhibitory activities, with the results indicating significant differences in receptor affinities between the two homologues. The analyses used included radioligand binding studies and a calcium fluorescence assay to determine the inhibitory activity of the compounds at the dopamine receptors.
10.1080/00397919308011121
The study presents a practical and convenient two-step synthesis of the title compound, 4,6-dichloro-5-benzylthiopyrimidine (3), starting from 4,6-dihydroxypyrimidine (1). The initial three-step approach involved converting 4,6-dihydroxypyrimidine (1) to 4,6-dihydroxy-5-bromopyrimidine (4) with an 80% yield. Then, compound (4) was reacted with benzylmercaptan and anhydrous potassium carbonate in N,N-dimethylformamide, yielding 4,6-dihydroxy-5-benzylthiopyrimidine (2) with a variable yield, the best being 50%. Finally, compound (2) was converted to the title compound (3) by heating in phosphorus oxychloride, resulting in a 75% yield. An improved method was developed using sulfenyl chloride chemistry, where benzyl sulfenyl chloride was prepared from dibenzyl disulfide and sulfuryl chloride, and reacted with 4,6-dihydroxypyrimidine (1) in N,N-dimethylformamide to obtain compound (2) in quantitative yield. The title compound (3) was then synthesized from compound (2) using phosphorus oxychloride, achieving an overall yield of approximately 72%. The study provides a more efficient and reliable synthesis route for 4,6-dichloro-5-benzylthiopyrimidine, which is a key precursor in the synthesis of various types of 4,6-di-substituted pyrimidine-5-sulfonamides with interesting biological activities.
10.1016/j.ejmech.2016.06.014
The research focuses on the synthesis and antimicrobial activity of a novel class of mono and bis heterocycles, including styryl, pyrrolyl, and pyrazolyl sulfonylmethyl-1,3,4-oxadiazolyl/thiadiazolyl amines. The study utilizes Z-styrylsulfonylacetic acid as a synthetic intermediate and employs various synthetic methodologies to prepare these compounds. The antimicrobial activity of these synthesized compounds was then evaluated against different bacterial and fungal strains. The reactants used in the synthesis encompass semicarbazide, thiosemicarbazide, POCl3, tosylmethyl isocyanide, sodium hydride, diazomethane, and chloranil, among others. The synthesized compounds were characterized using techniques like infrared (IR) spectroscopy, nuclear magnetic resonance (NMR), high-resolution mass spectrometry, and elemental analysis. The antimicrobial activity was assessed using the agar well diffusion method and broth dilution test to determine the minimum inhibitory concentration (MIC), minimum bactericidal concentration (MBC), and minimum fungicidal concentration (MFC). The findings revealed that mono heterocyclic compounds, particularly 5-(4-chlorostyrylsulfonylmethyl)-1,3,4-thiadiazol-2-amine (5c), exhibited superior antimicrobial activity against certain bacteria and fungi compared to the bis heterocyclic systems.
10.1016/j.tetlet.2006.02.090
The research focuses on the synthesis of novel heterocyclic systems, specifically imidazo[2,1-b][1,3,4]thiadiazole and imidazo[2,1-b][1,3]thiazole fused diazepinones, through an intramolecular amidation reaction. The study explores the ring opening and cyclization of specific precursors, which is a significant task in medicinal chemistry due to the therapeutic properties of these compounds. The reactants used include 2-amino-5-alkyl/aryl-1,3,4-thiadiazole, 3-(bromoacetyl)coumarin, and other substituted imidazothiadiazoles and imidazothiazoles. POCl3 (Phosphoryl chloride) was employed in the Vilsmeier–Haack reaction to facilitate the formylation of the thiadiazole derivatives, converting them into aldehydes. The synthesis process involves reactions such as the Vilsmeier–Haack formylation and intramolecular nucleophilic attack. The analyses used to characterize the synthesized compounds include infrared (IR) spectroscopy, proton nuclear magnetic resonance (1H NMR), carbon-13 nuclear magnetic resonance (13C NMR), and elemental analysis, which provide information on the structure, functional groups, and composition of the products. The research presents a methodology for creating new heterocyclic systems using readily available precursors and mild conditions.
10.1016/j.ejmech.2013.10.058
The research focuses on the synthesis and evaluation of novel 2-chloro-4-anilino-quinazoline derivatives as dual inhibitors of EGFR (Epidermal Growth Factor Receptor) and VEGFR-2 (Vascular Endothelial Growth Factor Receptor 2), which are established targets in cancer therapy. The study aims to develop compounds that can synergistically enhance antitumor activity and prevent resistance. The experiments involved the synthesis of various 2-chloro-4-anilino-quinazoline derivatives, utilizing key intermediates such as 2,4-dichloro-quinazolines. Phosphorus Oxychloride (POCl3) was used in the synthesis of 2,4-dichloro-quinazolines from quinazolinediones. N,N-Diisopropylethylamine (DIPEA) was used as a base in the nucleophilic aromatic substitution reactions. The synthesized compounds were then tested for their inhibitory effects on EGFR and VEGFR-2 using a radiometric protein kinase assay. Additionally, molecular docking studies were conducted to understand the molecular interactions of these compounds with the kinase domains of EGFR and VEGFR-2. The results identified compound 8o as particularly potent, showing approximately 7-fold and 11-fold increased inhibition potency against VEGFR-2 and EGFR, respectively, compared to the prototype compound.
10.1039/b710231c
The research describes the synthesis and biochemical evaluation of O-acetyl-ADP-ribose (OAADPr) and its N-acetyl analogs, which are non-hydrolyzable compounds designed to interact with the macro domain of histone protein H2A1.1. The purpose of this study was to develop a synthetic pathway for OAADPr and its analogs to overcome the limitations posed by the inherent instability and scarcity of enzyme-derived OAADPr, which is crucial for evaluating its cellular function and as a biochemical tool. The researchers successfully synthesized OAADPr and two N-acetyl analogs, demonstrating that these analogs are stable and can mimic OAADPr in binding with macroH2A1.1, suggesting their potential as valuable tools for future studies on the physiological role of OAADPr and for uncovering unidentified targets. The chemicals used in the process included various reagents such as benzyl bromide, sodium borohydride, acetic anhydride, pyridine, phosphorus oxychloride, and triethylamine, among others, to achieve the desired synthetic products through a series of chemical reactions and purification steps.

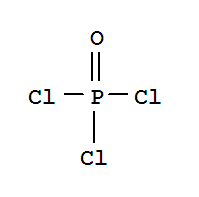

 T+,
T+, C
C






