ACS Catalysis
Research Article
entry 3, less than 5 turnovers were observed after 4 h. In
contrast, 3 and 5 provided 340 and 430 turnovers under these
conditions at 70 °C (entries 4−5). The efficiency of 6 as a
catalyst for hydrogenation at higher temperatures (fast
exchange conditions), but not at lower temperatures (slow
exchange conditions), suggests that the reversible binding of
ASSOCIATED CONTENT
Supporting Information
Crystallographic data in CIF format. Further details are given in
■
*
S
CO is likely relevant to catalysis by 6 at elevated temperatures.
2
AUTHOR INFORMATION
Notes
■
*
Importantly, the reversible formation of 3 from 6 under the
conditions for catalysis does not rule out the possibility of
direct CO2 hydrogenation at 6 (catalytic cycle shown in
Scheme 6). To explore this latter possibility, we first examined
t
The authors declare no competing financial interest.
the stoichiometric reaction of 6 with 1 equiv of KO Bu in
dimethylsulfoxide (DMSO) at 25 °C. After 5 min, a color
change from yellow to bright orange was observed,
accompanied by the complete conversion of 6 to a new Ru−
H species, 7 (Scheme 7). This complex proved challenging to
ACKNOWLEDGMENTS
■
This work was supported by the U.S. National Science
Foundation under the CCI Center for Enabling New
Technologies Through Catalysis (CENTC) Phase II Renewal,
CHE-1205189. C.A.H. was supported by a NSF Graduate
Research Fellowship and by a Rackham Merit Fellowship. We
also acknowledge funding from NSF Grant CHE-0840456 for
X-ray instrumentation and the contribution of Dr. Jeff Kampf in
collecting and refining crystallographic data. Finally, we thank
Dr. Alex Miller (UNC) and Dr. Karen Goldberg (UW) for
helpful discussions.
Scheme 7. Formation of Anionic Ru Complex 7 by
Deprotonation of 6
REFERENCES
■
(
1) Reutemann, W.; Kieczka, H. Formic Acid. In Ullmann’s
Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany,
35
isolate in high purity, as it is extremely moisture sensitive;
however, an in situ-generated sample of 7 was fully
characterized by H and C NMR spectroscopy.
2011.
1
13
(2) Schaub, T.; Paciello, R. A. Angew. Chem., Int. Ed. 2011, 50, 7278.
(3) Wesselbaum, S.; Hintermair, U.; Leitner, W. Angew. Chem., Int.
Ed. 2012, 51, 8585.
To probe whether 7 can participate in steps ii and iii of the
catalytic cycle proposed in Scheme 6, a sample of 7 was
35
(4) Zhang, Z.; Xie, Y.; Li, W.; Hu, S.; Song, J.; Jiang, T.; Han, B.
heated in DMSO-d in the presence of 1 bar of a 4:1 mixture of
6
Angew. Chem., Int. Ed. 2008, 47, 1127.
H :CO at 120 °C for 1 h in the absence of exogenous base
2
2
(
(
(
5) Leitner, W. Angew. Chem., Int. Ed. 1995, 34, 2207.
6) Jessop, P. G.; Ikariya, T.; Noyori, R. Chem. Rev. 1995, 95, 259.
7) Jessop, P. G. Homogeneous Hydrogenation of Carbon Dioxide.
(
Scheme 8). Under these conditions, 15% yield of HCOOK
Scheme 8. Stoichiometric Conversion of CO to Formate
2
In Handbook of Homogeneous Hydrogenation; Wiley-VCH: Weinheim,
Germany, 2007; p 489.
with 7
(
8) Jessop, P. G.; Joo, F.; Tai, C.-C. Coord. Chem. Rev. 2004, 248,
425.
9) Wang, W.; Wang, S.; Ma, X.; Gong, J. Chem. Soc. Rev. 2011, 40,
703.
10) Jeletic, M. S.; Mock, M. T.; Appel, A. M.; Linehan, J. C. J. Am.
Chem. Soc. 2013, 135 (31), 11533−11536.
11) Langer, R.; Disken-Posner, Y.; Leitus, G.; Shimon, L. J. W.; Ben-
David, Y.; Milstein, D. Angew. Chem., Int. Ed. 2011, 50, 9948.
12) Federsel, C.; Ziebart, C.; Jackstell, R.; Baumann, W.; Beller, M.
Chem.Eur. J. 2012, 18, 72.
13) Federsel, C.; Boddien, A.; Jackstell, R.; Jennerjahn, R.; Dyson, P.
J.; Scopelliti, R.; Laurenczy, G.; Beller, M. Angew. Chem., Int. Ed. 2010,
9, 9777.
14) Ziebart, C.; Federsel, C.; Anbarasan, P.; Jackstell, R.; Baumann,
2
(
3
(
(
(
3
6
was detected. This result suggests that the 6-catalyzed
(
hydrogenation of CO (Scheme 6) is a potentially viable
route to formate, albeit a likely minor pathway relative to that
depicted in Scheme 3.
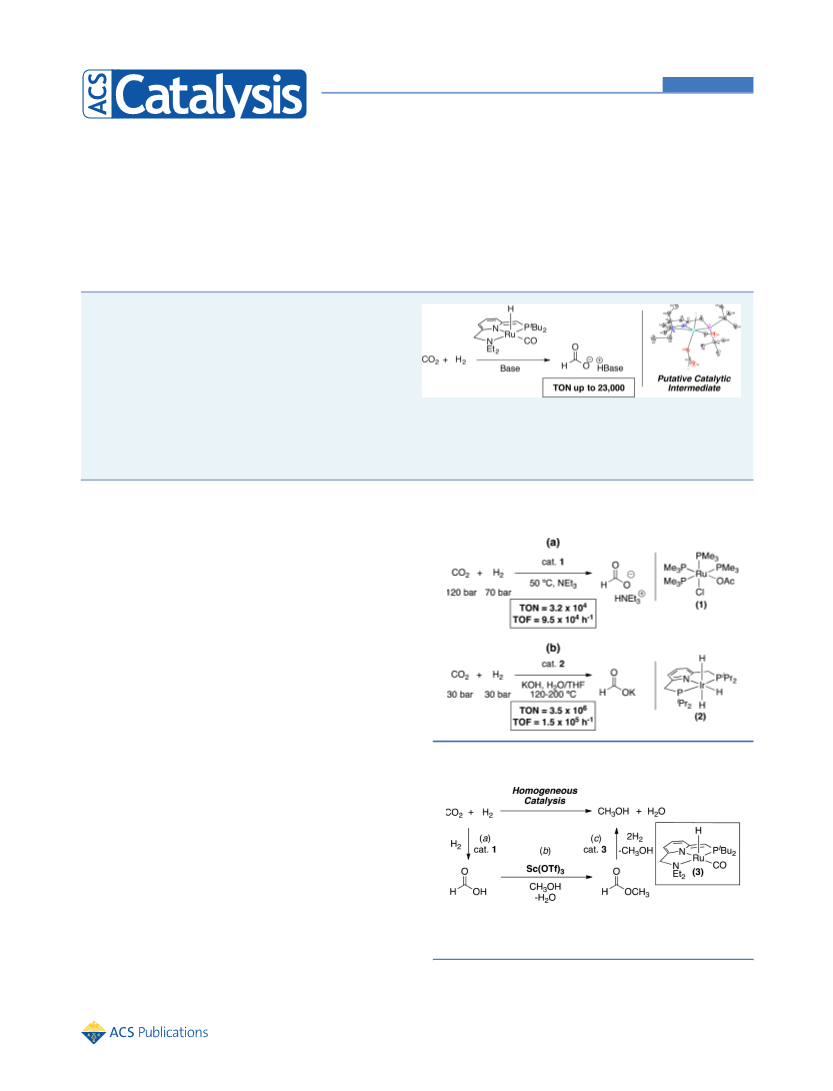
In summary, this paper has shown that Ru(PNN)(CO)(H)
3), a known ester hydrogenation catalyst, can also catalyze the
hydrogenation of CO to formate in the presence of a base. The
transformation is proposed to proceed through a mechanism
involving (i) heterolytic cleavage of H at 3 to form a Ru−
dihydride species, (ii) CO insertion to generate a Ru−formate
complex, and (iii) base-promoted release of formate. The
feasibility of each of these proposed mechanistic steps has been
demonstrated through stoichiometric studies of organometallic
intermediates. Ongoing work in this area aims to exploit this
newfound reactivity of 3, in combination with its ester
hydrogenation activity, to accomplish 3-catalyzed production
of MeOH from CO2.
2
4
(
W.; Spannenberg, A.; Beller, M. J. Am. Chem. Soc. 2012, 134, 20701.
(15) Huff, C. A.; Sanford, M. S. J. Am. Chem. Soc. 2011, 133, 18122.
(16) Wesselbaum, S.; vom Stein, T.; Klankermayer, J.; Leitner, W.
Angew. Chem., Int. Ed. 2012, 51, 7499.
(
2
(
17) Munshi, P.; Main, A. D.; Linehan, J. C.; Tai, C.-C.; Jessop, P. G.
J. Am. Chem. Soc. 2002, 124, 7963.
18) Tanaka, R.; Yamashita, M.; Nozaki, K. J. Am. Chem. Soc. 2009,
31, 14168.
19) PNN = 6-(di-tert-butylphosphinomethylene)-2-(N,N-diethyla-
minomethyl)-1,6-dihydropyridine).
20) Zhang, J.; Leitus, G.; Ben-David, Y.; Milstein, D. J. Am. Chem.
2
2
(
1
(
(
Soc. 2005, 127, 10840.
(21) Zhang, J.; Leitus, G.; Ben-David, Y.; Milstein, D. Angew. Chem.,
Int. Ed. 2006, 45, 1113.
2
415
dx.doi.org/10.1021/cs400609u | ACS Catal. 2013, 3, 2412−2416

 Huff, Chelsea A.
Huff, Chelsea A.