ꢁ
model aliphatic acidic compound undergoing direct minerali-
sation to CO2 and H2O.19,20
of ClO4 anions for TiO2 and their low reactivity towards
hydroxyl radicals.25 No pH change was noticed during the
runs under such conditions. By contrast, the pH value of
suspensions containing formic acid and benzoic acid increased
during the runs, from 3.5 to 5.8 and from 4.2 to ca. 6.0,
respectively, as a direct consequence of the mineralisation of
the acids to CO2 and H2O.26 With these acidic substrates, the
runs were performed under so-called natural pH conditions,
corresponding to the range of maximum fluoride adsorption
on TiO2, i.e. no buffer was added to the suspensions, to avoid
possible interference of other species (mainly anions) on the
photoredox processes at the TiO2–water interface.
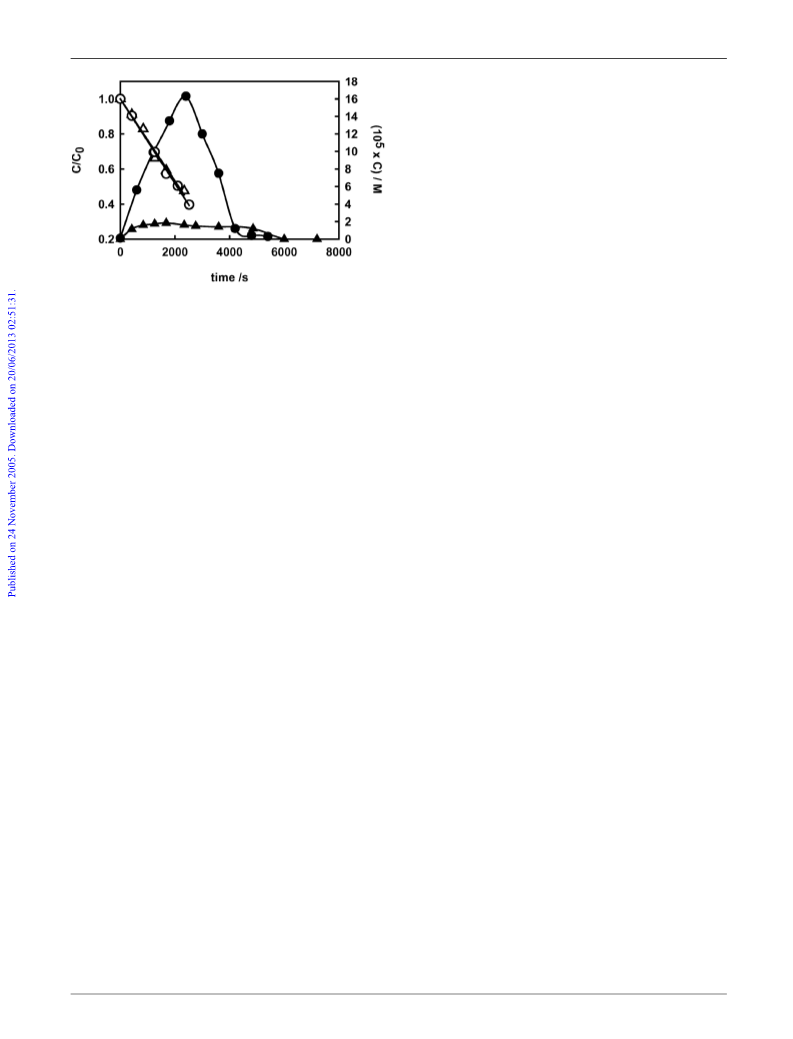
The origin of the peculiarly outstanding amount of H2O2
evolved during FA photocatalytic degradation on F–TiO2 was
clarified by investigating the effects of the addition of thꢁe
ꢀ
nitrate anion, which evidenced the central role of the CO2
species, formed by photoinduced FA oxidation on the photo-
catalyst surface.
Experimental
Materials
2-mL samples were periodically withdrawn from the reactor
and analysed, after removal of TiO2 particles by centrifugation
at 3000 rpm for 30 min, employing an ALC 4225 centrifuge.21
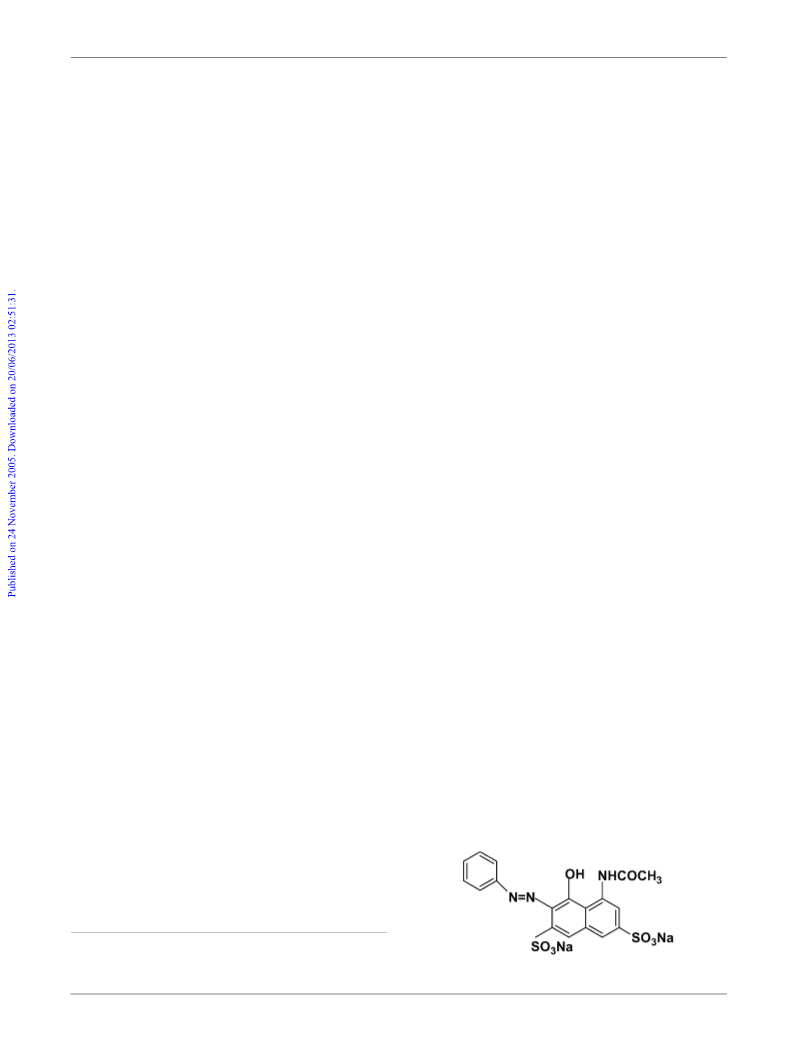
The cleavage of the azo bond of AR1, leading to its bleaching
(also mentioned as AR1 degradation), was monitored by
spectrophotometric analysis at 531 nm (maximum AR1 ab-
sorption, e = [3.13 ꢃ 0.02] ꢄ 104 Mꢁ1 cmꢁ1) by means of a
Perkin Elmer Lambda 16 spectrophotometer.21 BA concentra-
tion during the runs was detected by HPLC analysis, employ-
ing an Agilent 1100 Series apparatus, equipped with a
mBondapack-C18 column and a UV-Vis detector. An acetoni-
trile : water 60 : 40 mobile phase was used for BA analysis,27
flowing at 1.0 mL minꢁ1. FA concentration changes were
detected using a total organic carbon (TOC) analyser in the
not purgeable organic carbon (NPOC) mode (Shimadzu In-
struments, TOC-5000A). All runs were repeated at least twice
to check their reproducibility.
Acid Red 1 (AR1), purchased from Aldrich, was purified by
repeated crystallisation from methanol. Its purity from organ-
ic contaminants was verified by NMR analysis. Degussa P25
titanium dioxide (mainly anatase) was employed as photo-
catalyst. Formic acid (FA, purity 95–97%), benzoic acid (BA,
purity >99.5%), 2-propanol (purity 99.5%), KNO3 (purity
99.5%) and NaF (purity 99.99%) were purchased from Al-
drich and employed as received. Water purified by a Milli-Q
water system (Millipore) was used throughout.
Apparatus
All degradation runs were carried out at (35 ꢃ 1) 1C under
atmospheric conditions in a magnetically stirred 400 mL
cylindrical Pyrex reactor, employing an experimental set up
similar to that already described.21 Illumination was per-
formed through the reactor Pyrex walls by means of a 250 W
iron alogenide lamp (Jelosil, model HG 200), emitting in the
315–400 nm wavelength range, with a mean emission intensity
Hydrogen peroxide concentration was monitored during the
photodegradation runs by fluorimetric analysis (lex = 316.5
nm, lem = 408.5 nm) of the fluorescent dimer formed in the
horseradish peroxidase-catalysed reaction of hydrogen perox-
ide with p-hydroxyphenylacetic acid,28,29 using a 605-10S
Perkin Elmer fluorescence spectrophotometer. H2O2 standard
solutions employed in calibration were analysed iodometri-
cally. The H2O2 concentration profiles obtained during BA
photocatalytic degradation were corrected for the relatively
small fluorescence signal originating from salicylic acid, one of
the first degradation intermediates of BA. The signal was
measured in blank photocatalytic runs under conditions iden-
tical to those employed in H2O2 analysis, but with no
p-hydroxyphenylacetic acid and enzyme addition.
on the reactor of 3.5 ꢄ 10ꢁ4 einstein Lꢁ1
s
ꢁ1, as periodically
checked by ferrioxalate actinometry.22
Procedures
The irradiated aqueous suspensions contained 0.1 g Lꢁ1 of
TiO2; the initial concentration (C0) of the substrates was 2.5 ꢄ
10ꢁ5 M for AR1, 1.0 ꢄ 10ꢁ4 M for BA and 5.0 ꢄ 10ꢁ4 M for
FA. Titanium dioxide fluorination was achieved by adding
0.01 M NaF, corresponding to 0.1 mol of Fꢁ per gram of
TiO2. Fluoride ions, able to very quickly displace the –OH
groups on the surface of titanium dioxide, were added to the
suspensions immediately before starting irradiation, to mini-
mise the coagulation of the photocatalyst.8,10 When investigat-
ing the effect of nitrate addition, the KNO3 concentration was
0.05 M, high enough to guarantee a quantitative scavenging of
the carbon dioxide radical anion.23
Adsorption studies were performed both in the presence and
in the absence of 0.01 M NaF, on suspensions containing 1.0 g
L
ꢁ1 of TiO2 and 2.5 ꢄ 10ꢁ5 M AR1, 1.0 ꢄ 10ꢁ4 M BA or 5.0 ꢄ
10ꢁ4 M FA. After continuous stirring for 24 h in the dark at
35 1C, the photocatalyst was removed and the liquid phase was
analysed for AR1, BA or FA residual content. The Fꢁ/TiO2
ratio employed in the adsorption experiments, lower with
respect to that of the photocatalytic degradation runs, guar-
anteed a similar displacement of surface –OH groups, the
maximum value of adsorbed fluoride being 2.5 ꢄ 10ꢁ4 mol gꢁ1
of TiO2.30
The pH was monitored during the runs by means of an
Amel Instruments 334-B pH-meter. A decrease in pH was
observed during AR1 photocatalytic degradation under nat-
ural pH conditions, from an initial value of 5.8 to a final value
of ca. 4.4, as a consequence of the production of stable acids,
due to the fast removal of sulfonic groups and the oxidation of
the azo double bond.24 Thus, to guarantee an efficient adsorp-
tion of fluoride anions on the surface of the oxide, during AR1
photodegradation the pH was lowered to 3.7 by adding small
amounts of HClO4, which is known to have negligible influ-
ence on the photocatalytic activity, because of the low affinity
The reduction potentials of AR1, BA and FA were deter-
mined by cyclic voltammetry measurement using a glassy
carbon working electrode vs. Ag/AgCl in 0.1 M HClO4 for
AR131 or 0.5 M NaClO4 for BA and FA solutions, with a scan
speed of 50 mV sꢁ1
.
ꢂc
This journal is the Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2006
New J. Chem., 2006, 30, 108–114 | 109

 Mrowetz, Marta
Mrowetz, Marta