The Journal of Organic Chemistry
Article
The studied mechanisms of DHBQ degradation by hydrogen
peroxide suggest that both reaction rate and reaction
mechanism (and thus product distribution) are controlled by
the pH since the underlying (de)protonation processes change
the stability and the reactivity of the biradical formed by the
O−O bond homolysis of the respective intermediates. We hope
that the insights will be helpful with regard to improving the
efficiency of pulp bleaching processes.
ASSOCIATED CONTENT
Supporting Information
■
*
S
Complete ref 13. C NMR and 11B NMR spectra of the
13
degradation reaction at 298.15 K and 240 s, sample A in
DMSO-d . Total-ion chromatogram in GC−MS analysis (after
6
trimethylsilylation) of degradation reaction at 298.15 K and 240
s, sample A. Relationships between ln[DHBQ] and t in the
degradation of DHBQ under the presence of sodium sulfate
and sodium chloride. Arrhenius plot in the degradation of
DHBQ under the presence of sodium sulfate. Important
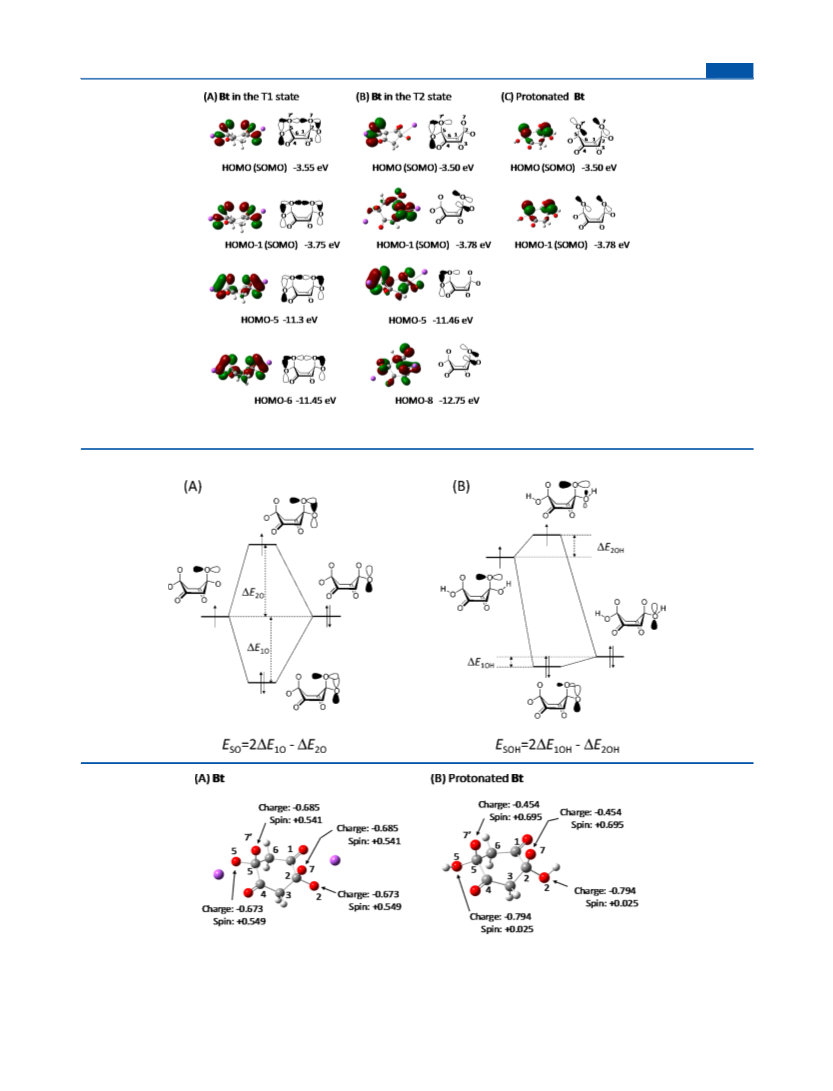
orbitals of Bs. Picture of the reaction solution under the pH 10
conditions. Geometry changes and energy profile in the ionic
degradation of DHBQ. Cartesian coordinates of the optimized
EXPERIMENTAL AND COMPUTATIONAL DETAILS
■
Experimental Section. All chemicals were purchased from
commercial providers. They were of highest purity available (p.a.
grade) and were used without further purification. Proton nuclear
1
magnetic resonance ( H NMR) spectra were recorded at 400.13 MHz
proton resonance frequency.
The degradation reaction was started by adding a 30% aqueous
solution of hydrogen peroxide (2.2 mL, 19.6 mM of hydrogen
peroxide) to 20 mL of an pH 10 buffer solution (NaOH-borax) of
DHBQ (10.7 mg, 0.0763 mM) in a 50 mL round-bottom flask. The
buffer solution of DHBQ contained 4.0 mg of 1,3,5-tricarboxylbenzene
as internal standard. The solution of DHBQ was preheated at
temperatures between 278.15 and 323.15 K before addition of the
hydrogen peroxide solution. After the reaction started, sampling of the
reaction mixture was done by adding 2.0 mL of the reaction solution
separately into 10.0 mL of a 0.005 M HCl solution and 10.0 mL of
deionized water, to obtain two types of the sample: sample A is
prepared from the 0.005 M HCl solution, and sample B from the pure
water. These samples were then immediately cooled to 0 °C in an ice
AUTHOR INFORMATION
Notes
■
*
The authors declare no competing financial interest.
ACKNOWLEDGMENTS
■
We performed quantum chemical calculations with the
workstation in the Sakaki group, Fukui institute for
fundamental chemistry at Kyoto University, Japan and are
thankful for the access. The financial support of the Austrian
Christian Doppler Research Society (CDG) through the CD-
lab “Advanced cellulose chemistry and analytics” and of the
Austrian Research Promotion Agency (FFG, project 829443) is
gratefully acknowledged.
bath and frozen at 193.15 K. After freeze-drying of the samples at this
1
temperature, sample A was analyzed by H NMR in DMSO-d to
6
quantify the amount of nonreacted DHBQ. Sample B was monitored
1
with H NMR in D O. Since sample B is not neutralized with HCl
2
during its preparation, all acids produced from DHBQ exist as their
corresponding sodium salts, which are not removed in the freeze-
drying process.
REFERENCES
The degradation of DHBQ was also carried out in the presence of
■
1
.63 g (11.4 mM) of sodium sulfate. In this case, sodium sulfate was
(1) (a) Rosenau, T.; Potthast, A.; Milacher, W.; Hofinger, A.; Kosma,
P. Polymer 2004, 45, 6437. (b) Rosenau, T.; Potthast, A.; Milacher, W.;
Adorjan, I.; Hofinger, A.; Kosma, P. Cellulose 2005, 12, 197.
(c) Rosenau, T.; Potthast, A.; Kosma, P.; Suess, U.; Nimmerfroh, N.
Holzforschung 2007, 61, 656. (d) Krainz, K.; Potthast, A.; Suess, U.;
Dietz, T.; Nimmerfroh, N.; Rosenau, T. Holzforschung 2009, 63, 647.
(e) Rosenau, T.; Potthast, A.; Krainz, K.; Yoneda, Y.; Dietz, T.;
Shields, Z. P.; French, A. D. Cellulose 2011, 18, 1623.
added into the buffer solution of DHBQ in advance, and the
degradation was started in the same manner as described above.
Sampling was carried out according to the procedure described above
for sample A. The freeze-dried mixture was extracted with dimethyl
1
ether, and the soluble part was subject to H NMR analysis in DMSO-
d6 after removal of the solvent in vacuo.
Computations. We employed the GAUSSIAN 09 program
13
packages for the calculations. The geometry optimization in water
(2) Hosoya, T.; French, A. D.; Rosenau, T. Mini-Rev. Org. Chem.
2013, 10, 309.
1
4
was carried out by the DFT method with the B3LYP functional and
the PCM method. The 6-31G(d) basis sets were employed for H, C,
O, Na where a diffuse function was added to each of C and O and a p-
polarization function was added to H. We name these basis sets as BS-
I. For single point calculations, we employed MP2 or DFT(UB3LYP)
method, the latter was used only for the calculations of I, Bs, and Bt.
The DFT(UB3LYP) functional is adequate to evaluate the property of
(3) (a) Brassard, P.; L’Ecuyer, P. Can. J. Chem. 1958, 36, 1346.
(b) Nicolaides, D. N.; Gautam, D. R.; Litinas, K. E.; Papamehael, T. J.
Chem. Soc., Perkin Trans. 1 2002, 1455. (c) Lang, M.; Muhlbauer, A.;
Jagers, E.; Steglich, W. Eur. J. Org. Chem. 2008, 20, 3544. (d) Shaabani,
A.; Ghadari, R.; Sarvary, A.; Rezayan, A. J. Org. Chem. 2009, 74, 4372.
(e) Shaabani, A.; Ghadari, R.; Ghasemi, S.; Pedarpour, M.; Rezayan,
A.; Sarvary, A.; Ng, S. W. J. Comb. 2009, 11, 956. (f) Misiolek, A.;
Ichimura, A. S.; Gentner, R. A.; Huang, R. H.; McCaffrey, V. P.;
Jackson, J. E. Inorg. Chem. 2009, 48, 9005. (g) Jimenez-Alonso, S.;
Estevez-Braun, A.; Ravelo, A. G.; Zarate, R.; Lopez, M. Tetrahedron
2007, 63, 3066. (h) Jimenez-Alonso, S.; Perez-Lomas, A. L.; Estevez-
Braun, A.; Martinez, F. M.; Orellana, H. C.; Ravelo, A. G.; Gamarro,
F.; Castanys, S.; Lopez, M. J. Med. Chem. 2008, 51, 7132. (i) Koulouri,
S.; Malamidou-Xenikaki, E.; Spyroudis, S. Tetrahedron 2005, 61,
10894.
(4) (a) Manthey, M. K.; Pyne, S. G.; Truscott, R. J. W. Aust. J. Chem.
1989, 42, 365. (b) Ikeda, M.; Kitahara, K.; Nishi, H. J. Heterocycl.
Chem. 1992, 29, 289. (c) Zhang, D.; Jin, G. X. Organometallics 2003,
22, 2851. (d) Gellerman, G.; Rudi, A.; Kashman, Y. Tetrahedron 1994,
50, 12959. (e) Reuben, G.; Shonle, H. A. J. Am. Chem. Soc. 1946, 68,
2246. (f) Placin, F.; Clavier, G.; Najera, F.; Desvergne, J. P.; Pozzo, J.
L. Polycyclic Aromat. Compd. 2000, 19, 107. (g) Lehaire, M. L.;
5
O−O bonds as we reported before, while the MP2 method did not
work due to the instability of the wave function that arises from the
homolysis of the O−O bond. In the single point calculations, cc-pVDZ
basis stets were employed for H and Na, and aug-cc-pVDZ basis sets
were employed for C and O. These basis sets are named as BS-II.
In all calculations, the solvation energy was evaluated with the PCM
method. For the determination of the cavity size in the PCM
calculation, the UFF parameters were used along with the united atom
topological model optimized for the HF/6-31G(d) level of theory for
geometry optimization and energy evaluation, respectively. It was
ascertained that each equilibrium geometry exhibited no imaginary
frequency and each transition state exhibited one imaginary frequency.
Enthalpy, entropy, and Gibbs energy changes were evaluated at 298.15
K. Zero-point energy, thermal energy, and entropy change were
evaluated with the DFT(B3LYP) method. The translational entropy in
15
water was evaluated according to the literature method.
1
1202
dx.doi.org/10.1021/jo401486d | J. Org. Chem. 2013, 78, 11194−11203

 Hosoya, Takashi
Hosoya, Takashi