228
M. Grigoras et al. / Dyes and Pigments 113 (2015) 227e238
Furthermore, we intend to extend this method to poly-
CV of ferrocene as the internal standard in an identical cell without
any compound in the system (E1/2 ¼ 0.40 V versus the Ag/AgCl).
Prior to the each experiment, the Bu4NClO4 solutions were deox-
ygenated by passing dry nitrogen gas for 10 min. All measurements
were performed at room temperature (25 ꢀC) under nitrogen
atmosphere.
arylenevinylenes containing triphenylamine substituted in the free
para position with phenylethynyl- and 3-pyridylethynyl groups.
The electron-rich triple bond could be post-modified by: (a) addi-
tion of tetracyanoethylene [18e26], (b) cycloaddition of tetraphe-
nylcyclopentadienone [10], (c) transformation into a quinoxaline
acceptor group [11], while pyridyl group can be (d) protonated or
quaternized with alkyl bromides [27], with final effects on the main
chain conjugation length. Routes (a) and (d) will be explored to
tune the optical and electronic properties of PPVs. So, in this paper
we report the synthesis and spectroscopic and electrochemical
characterization of poly [4,4'-(400-phenylethynyl)-triphenylamine
vinylene] and poly[4,4'-(400-3-pyridylethynyl)-triphenylamine
vinylene] obtained by Stille polycondensation. A comparative
analysis of the optical and electronic properties of parent and
chemical modified polymers will be presented and correlated using
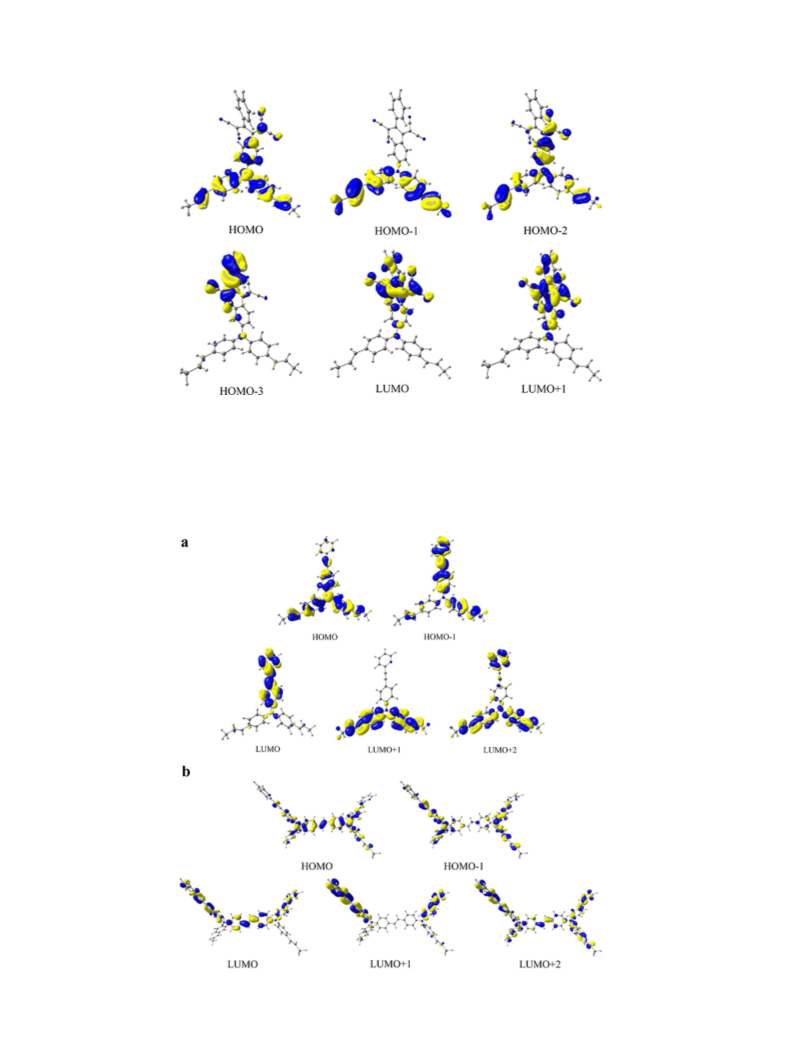
time-dependent density functional theory (TD-DFT) calculations.
The TD-DFT analysis can provide a link between structure and NMR,
absorption and luminescence spectra of such polymers [28,29].
2.3. Monomers and polymers synthesis
The synthetic routes for triphenylamine-based monomers
starting from triphenylamine, are outlined in Scheme 1. 4,4',400-
Trisiodotriphenylamine and 4,4'-diiodotriphenylamine (1) were
obtained by iodination of triphenylamine with KI/KIO3 in acetic
acid, using known methods [30,31]. The Sonogashira coupling of
4,4',400-trisiodotriphenylamine with phenylacetylene and 3-
pyridylacetylene in the presence of catalyst [(PPh3)2PdCl2, CuI,
PPh3] using triethylamine as solvent gave monomers 2 and 3.
2.3.1. Synthesis of bis(4-iodophenyl)-phenylamine (1)
In a 250 mL two-neck round bottom flask equipped with mag-
netic stirrer, condenser and nitrogen inleteoutlet were introduced
triphenylamine (8 g, 3.2 mmol), KI (7.19 g, 4.3 mmol), and glacial
acetic acid (120 mL). The mixture was stirred in nitrogen atmo-
sphere at 85 ꢀC for 5 h and KIO3 (4.60 g, 2.15 mmol) was introduced
over 5 h. The mixture was precipitated in water and a dirty white
compound was obtained and purified by column chromatography
using ethyl acetate/hexane (1:5) as eluent. Yield: 66%,
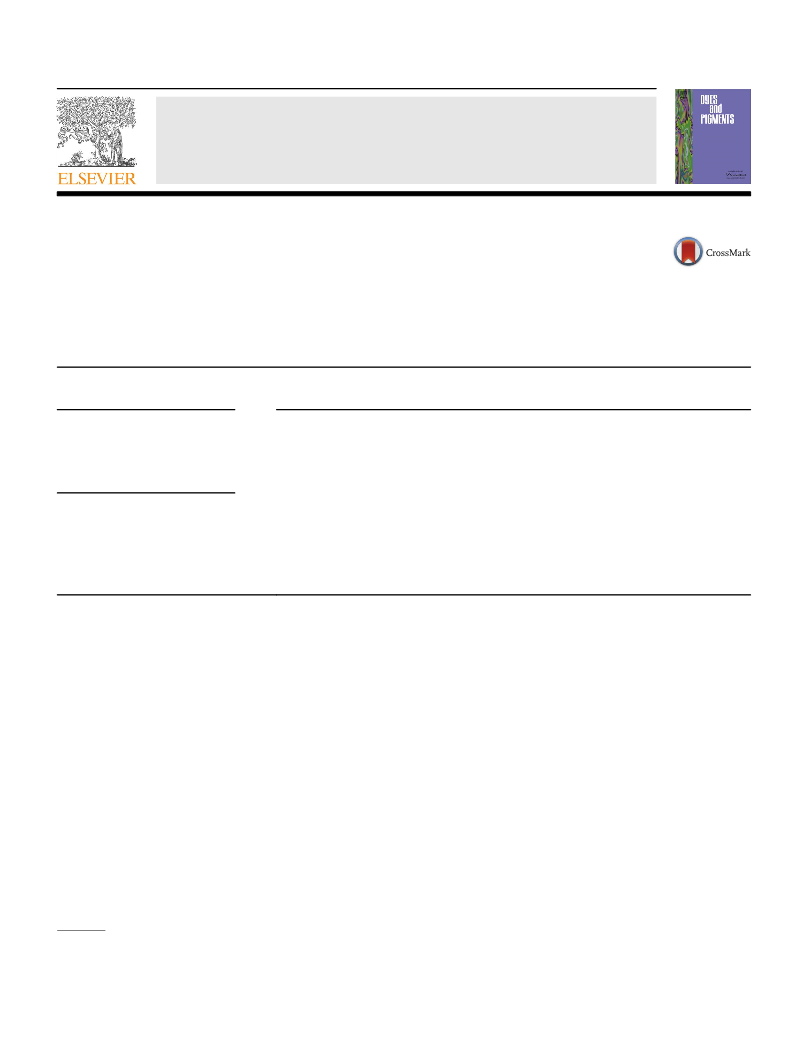
Mp ¼ 69e70 ꢀC. 1H NMR (CDCl3, ppm): 7.50 (4H, d, J ¼ 8.8 Hz), 7.24
(2H, d, J ¼ 8.0 Hz), 7.06e7.04 (3H, d þ t), 6.82e6.80 (4H, d,
J ¼ 8.8 Hz).
2. Experimental
2.1. Materials
Triphenylamine, 3-ethynylpyridine, triethylamine, Pd(PPh3)4,
PPh3, CuI, trans-1,2-bis(tributylstannyl)ethene, all from Aldrich and
phenylacetylene (Fluka), were used as received. Other reactants
and solvents are obtained from commercial sources and used as
received or dried by known methods. Tetrabutylammonium per-
clorate ((C4H9)4NClO4) was used as supporting electrolyte in elec-
trochemical studies. All manipulations were carried out under an
inert atmosphere using the Schlenk technique.
2.3.2. Synthesis of N,N-bis(4-iodophenyl)-4'-(phenylethynyl)
phenylamine (2)
2.2. Measurements
In a 100 mL three-neck round bottom flask equipped with
magnetic stirrer, condenser and nitrogen inleteoutlet were intro-
duced phenylacetylene (0.47 mL, 4.3 mmol), PdCl2$2PPh3 (0.011 g),
CuI (0.018 g), PPh3 (0.0165 g), TEA (8 mL) and reaction mixture was
stirred at room temperature for 2 h. 4,4',400-Trisiodotriphenylamine
(2.678 g, 4.3 mmol) dissolved in TEA (5 mL) was added to the flask
and the reaction mixture was maintained 24 h at 50e60 ꢀC. The
product was separated by precipitation in water, washed with
aqueous solution 0.1 M HCl and dried. Pure yellow crystalline
compound was obtained by flash chromatography using hexane as
eluent. Yield ¼ 88.4%. Mp ¼ 74e75 ꢀC. ESI-MS ¼ 598.2 (M þ Hþ). IR
(KBr, cmꢁ1): 3043, 2210, 1634, 1574, 1505, 1313, 1265, 1177, 1058,
1002, 912, 816,753, 689. 1H NMR (CDCl3, ppm): 7.58e7.48 (6H, m),
7.4 (2H, t), 7.35e7.30 (3H, m), 7.05e6.95 (2H, m), 6.9e6.75 (4H, m).
13C NMR (CDCl3, 100.39 MHz, ppm): 146.89, 135.98, 132.84, 132.6,
131.55, 128.39, 128.17, 126.13, 123.40, 123.11, 117.55, 116.37, 89.24,
89.22.
FT-IR spectra were recorded in KBr pellets on a DIGILAB-FTS
2000 spectrometer. 1H NMR and 13C NMR spectra were recorded
at room temperature on a Bruker Avance DRX-400 spectrometer
operating respectively at 400 MHz (for 1H) and 100.39 MHz
(for 13C) as solutions in CDCl3 or DMSO-d6 and chemical shifts are
reported in ppm and referenced to TMS as internal standard or
DMSO-d5. UVeVis and fluorescence measurements were carried
out in CHCl3 solution (spectrophotometric grade) or as a film on
quartz glass, on a Specord 200 spectrophotometer and Perkin Elmer
LS 55 apparatus, respectively. The fluorescence quantum yields
were determined by the integrating sphere method using an FLS
980 spectrometer and CHCl3 as solvent upon excitation with
lex ¼ 400 nm. Differential scanning calorimetry (DSC) and ther-
mogravimetric analyses (TGA) were carried our in a TGA/DTA STA
449 F1 Netzsch (Germany) at a heating rate of 10 ꢀC/min in a ni-
trogen flow. The relative molecular weights were determined by gel
permeation chromatography (GPC) using a PL-EMD 950 Evapora-
tive Mass Detector instrument and polystyrene standards for the
calibration plot and chloroform (1 mL/min) as solvent.
2.3.3. Synthesis of N,N-bis(4-iodophenyl)-4'-(3-pyridylethynyl)
phenylamine (3)
In a 50 mL three-neck round bottom flask equipped with mag-
netic stirrer, condenser and nitrogen inleteoutlet were introduced
3-ethynylpyridine (0.165 g, 1.6 mmol), PdCl2$2PPh3 (0.056 g,
0.08 mmol), CuI (0.03 g, 0.16 mmol), PPh3 (0.04 g, 0.16 mmol),
triethylamine (15 mL) and tetrahydrofuran (5 mL). The mixture was
stirred in nitrogen atmosphere at 50e60 ꢀC for an hour after that
4,4',400-trisiodotriphenylamine (1 g, 1.6 mmol) dissolved in a
mixture of TEA (5 mL) and THF (5 mL) was added. Then the mixture
was stirred at 80 ꢀC when the solution color turned out in time from
yellow to red and some solids are deposited on the flask's walls.
After 24 h the mixture was precipitated in water, filtrated and dried.
The cyclic voltammograms (CV) were recorded using a Bio-
analytical System, PotentiostateGalvanostat (BAS 100B/W). The
electrochemical cell was equipped with three electrodes: a working
electrode (disk shape Pt electrode,
F
¼ 1.6 mm), an auxiliary
electrode (platinum wire), and a reference electrode (consisted of a
silver wire coated with AgCl). Before experiments, Pt electrode was
polished between each set of experiments with aluminium oxide
powder on a polishing cloth, and then was sonicated in a mixture of
detergent and methanol for 5 min and then rinsed with a large
amount of doubly distilled water. The reference electrode (Ag/Agþ)
was calibrated at the beginning of the experiments by running the
The pure orange compound
3 was obtained by silicagel

 Grigoras, Mircea
Grigoras, Mircea