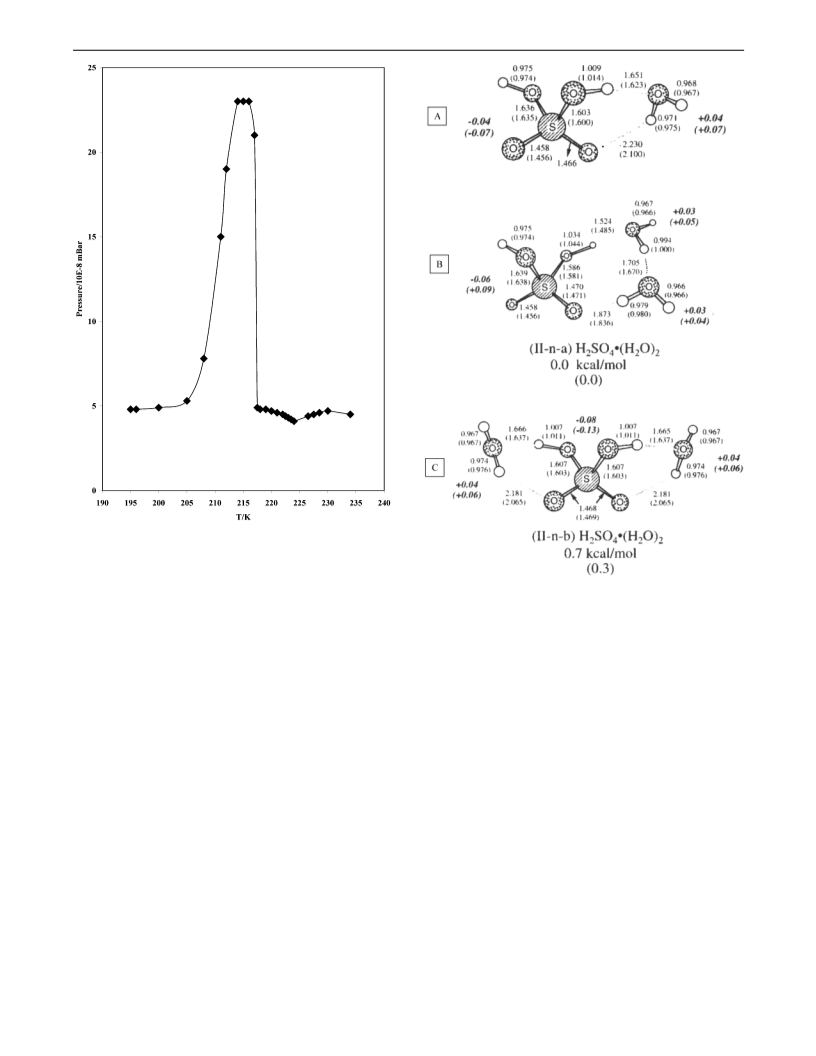
Identification of Molecular Sulfuric Acid Hydrates
A R T I C L E S
cally.6,7 Although there are some discrepancies, there is
reasonable agreement in the literature as to the nature of this
process. Computational studies suggest that the reaction of SO3
with one water molecule has an activation barrier of about 28
kcal mol-1, with the high energy barrier being due to a sterically
hindered four centered transition state. The introduction of a
second water molecule reduces this energy barrier to about 13
kcal mol-1 through the formation of a six centered transition
state. Additional water molecules continue to reduce the barrier
either by acting as microsolvent stabilizing the transition state
or by allowing alternative reaction mechanisms (i.e., stepwise
rather than concerted proton transfers). The presence of 10 or
more water molecules effectively removes the barrier for the
reaction. These predictions are supported by experimental work.
For example, the gas-phase SO3 + H2O reaction has been shown
experimentally by Jayne et al to be second order with respect
to the partial pressure of water and to have a strong negative
temperature dependence.7 In the pressure range from 133 to
1000 mbar and temperature range from 283 to 370 K, Jayne et
al observed Arrhenius behavior with A ) 3.90 × 10-41 cm-6
molecule-2 s-1 and Ea ) -13.5 kcal mol-1 for the reaction.
However, at lower temperatures (243-268 K) and at SO3
densities of ∼1012 molecules cm-3, very fast decay rates showed
a trend to first-order dependence in water vapor pressures, which
may represent an efficient heterogeneous reaction between SO3
and acid water particles at reduced temperature.
Theoretical studies predict reaction products which involve
molecular sulfuric acid complexed with varying amounts of
hydrogen bonded water in the gas phase. This behavior is
confirmed in molecular beam experiments, with the principal
product being an un-ionized monohydrate complex for which
detailed structural parameters have been obtained.8 In order for
the ionic species observed in condensed films and aerosols to
be formed, these complexes must further hydrate and coalesce
during the formation of an aerosol particle. In this paper,
spectroscopic observations of the molecular products of the
heterogeneous hydration of SO3 collected on a cold ATR
element are presented, along with an analysis of the effects of
temperature and water partial pressure upon the subsequent
formation of intermediates and ionic hydrates.
valves with internal dosing tubes are directed at the open faced
of the IRE; this enables co-dosing of reactive gases. The gases
for dosing are prepared in separate gas lines to prevent pre-
reaction, and films are deposited by simultaneous dosing of SO3
and H2O (or D2O). SO3 gas is prepared by sublimation of solid
SO3 (Aldrich, U.K.) into a preconditioned, evacuated gas line.
Deionized H2O or D2O (Aldrich, U.K.) is vacuum distilled and
degassed by repeated freeze/pump/thaw cycles.
SO3 and H2O are co-dosed on to the IRE between 190 and
200 K, which is above the deposition temperature for either ice
or SO3 at partial pressures of less than 10-5 mbar. The partial
pressures of the reacting gases are monitored during deposition
by a nude ion gauge and a residual gas analyzer. Calibration of
the absolute pressures at the sample IRE face is performed by
measuring the frost point of water vapor at a set temperature.
The local pressures at the surface of the IRE are calculated from
a knowledge of the relationship between the measured chamber
pressure and the local pressure at the exit points of the dosing
tubes.
Results
Formation and Structure of Amorphous Sulfuric Acid
Films. At 190 K, neither SO3 nor H2O form stable single
component films of appreciable thickness at partial pressures
of less than 1 × 10-5 mbar. The deposition of material upon
the IRE surface must therefore be due to either a rapid gas-
phase reaction or a heterogeneous process with one or both
components adsorbed prior to reaction. Extrapolating the
reaction rate for H2O(g) + SO3(g) determined by Jayne et al.7
down to 180 K gives a very long half-life for the reaction, and
since the mean free path at pressures below 1 × 10-4 mbar is
of the order of meters there is not a significant probability of
the reaction occurring in the gas phase in the short distance
between the ends of the dosing tubes and the face of the IRE.
In their kinetic study, Jayne et al observed that at low
temperatures and at relatively high SO3 pressures (>∼1012
molecules cm-3) the reaction is very fast due to a heterogeneous
reaction at the surface of the acid-water film. Further evidence
for the likelihood of a heterogeneous process in these experi-
ments comes from an observation during sample preparation.
The Ge IREs used in this laboratory are cleaned prior to use by
etching in dilute HF. This renders the surface extremely
hydrophobic. Upon subsequent surface oxidation (either chemi-
cally or by standing in the atmosphere), the IRE becomes
hydrophilic in nature. In vacuo uptake of water is observed to
be retarded on the fresh hydrophobic surface, and the reaction
between SO3 and H2O does not occur at pressures below 1 ×
10-5 mbar. However, reaction is observed immediately upon
exposure above the hydrophilic surface and a film of 5 nm
thickness is deposited in ca. 5 min. This behavior is attributed
to the immobilization of a thin (possibly monolayer) film of
H2O by interaction between gaseous H2O and surface oxide and
hydroxide functionalities which initiates the heterogeneous
process. In these experiments, the hydrophilicity of the IRE
surface increases the lifetime of surface-bound H2O and permits
the SO3(g) + H2O(ads) reaction to be studied in detail.
Experimental Section
The experimental apparatus used in this study has been
described previously.9 Briefly, a germanium internal reflection
element (IRE) is held on a thermostated sample mount in a high
vacuum chamber. The temperature of the IRE, measured by a
K type thermocouple, is controlled by the combination of liquid
nitrogen cooling and resistive heating. Three high precision leak
(6) (a) Hofman, M.; Schleyer, P.von R. J. Am. Chem. Soc. 1994, 116, 4947.
(b) Morukuma, K.; Muguruma, C. J. Am. Chem. Soc. 1994, 116, 10 316.
(c) Kolb, C. E.; Jayne, J. T.; Worsnop, D. R.; Molina, M. J.; Meads, R. F.;
Viggiano, A. A. J. Am. Chem. Soc. 1994, 116, 10 314. (d) Phillips, J. A.;
Canagaratna, M.; Goodfriend, H.; Leopold, K. R. J. Phys. Chem. 1995,
99, 501. (e) Lovejoy, E. R.; Hanson, D. R.; Huey, L. G. J. Phys. Chem.
1996, 100, 19 911. (f) Akhmatskaya, E. V.; Apps, C. J.; Hillier, I. H.;
Masters, A. J.; Palmer, I. J.; Watt, N. E.; Vincent, M. A.; Whitehead, J. C.
J. Chem. Soc., Faraday. Trans. 1997, 93, 2775. (g) Meyer, E. J.; Sprik,
M. J. Phys. Chem. A 1998, 102, 2893. (h) Loerting, T.; Liedl, K. R. Proc.
Nat. Acad. Sci. 2000, 97, 8874. (i) Loerting, T.; Kroemer, R. T.; Liedl, K,
R. Chem. Commun. 2000, 999. (j) Loerting, T.; Liedl, K. R., J. Phys. Chem.
A 2001, 105, 5137.
By variation of the ratio of the partial pressures of H2O and
SO3 above the IRE, the composition of the film which is
deposited on the cold IRE surface can be controlled. (NB, For
the purposes of simplicity, hereafter this ratio is defined as Rp,
(7) Jayne, J. T.; Po¨schl.; U, Chen, Y.; Dai, D.; Molina, L. T.; Worsnop, D. R.;
Kolb, C. E.; Molina, M. J. J. Phys. Chem. A. 1997, 101, 10 000.
(8) Fiacco, D. L.; Hunt, S. W.; Leopold, K. R. J. Am. Chem. Soc. 2002, 124,
4504.
(9) Horn A. B.; Sully, K. J. J. Chem. Soc.: Faraday Trans. 1997, 93, 2741.
9
J. AM. CHEM. SOC. VOL. 125, NO. 7, 2003 1995

 Couling, Suzanne B.
Couling, Suzanne B.