Organometallics
ARTICLE
155.60 (s, o-Py1), 152.5 (s, o-Py2), 149.51 (s, o-Py1 CH), 141.46 (s, o-Ar),
139.37 (s, p-Ar), 138.86 (s, p-Py1-CH), 136.86 (s, p-Py2-CH), 134.33
(s, Py-NCHN), 130.89 (s, Ar-C-N), 129.98 (s, m-ArCH), 124.34 (s,
m-Py1 CH), 124.13 (s, m-Py2 CH), 123.27 (s, imidazole-CHCHN),
122.54 (s, imidazole-CHCHN), 121.39 (s, m-Py1 CH), 120.96 (s, m-Py2
CH), 54.26 (s BPyCH2N), 21.18 (s, p-ArCH3), 17.68 (s,o-ArCH3). HRMS:
m/z 355.1915 (M+, calcd m/z 355.1923). Anal. Calcd for C23H23ClN4: C,
70.67; H, 5.93; N, 14.33. Found: C, 70.20; H, 5.87; N, 14.46
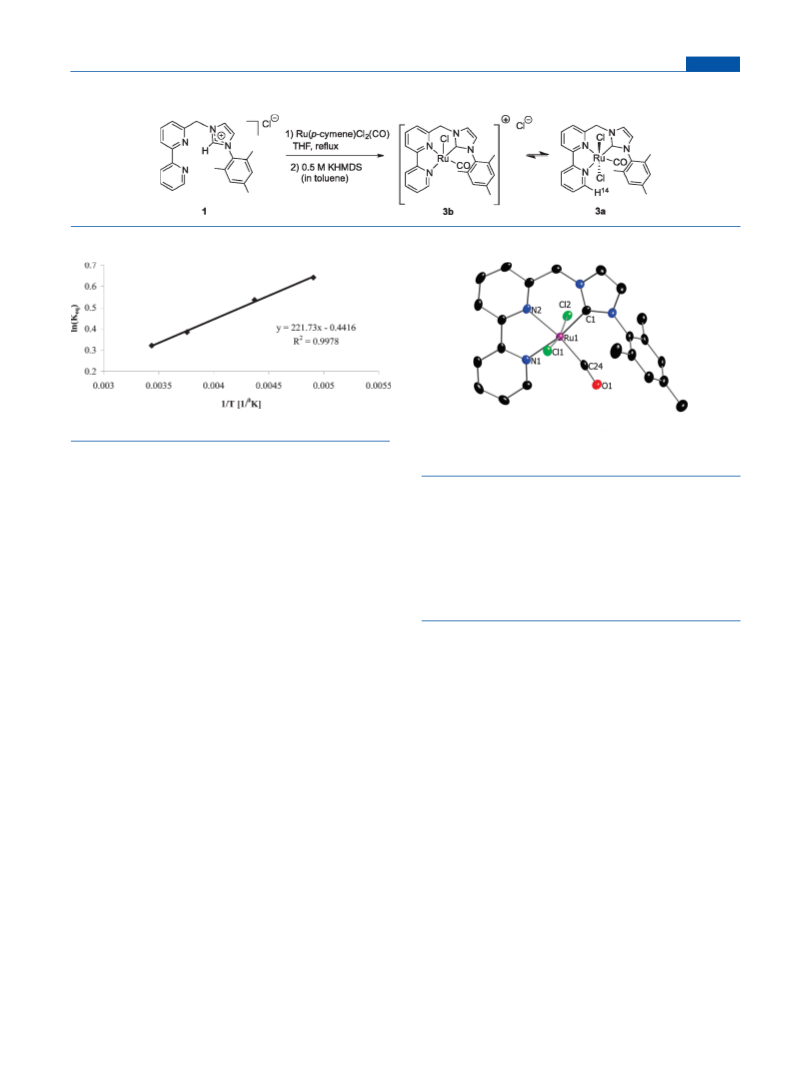
Table 5. Keq at Various Temperatures in CD2Cl2
3b h 3a
K
T (K)
1.9
203.86
228.76
266.12
291.02
1.71
1.47
1.38
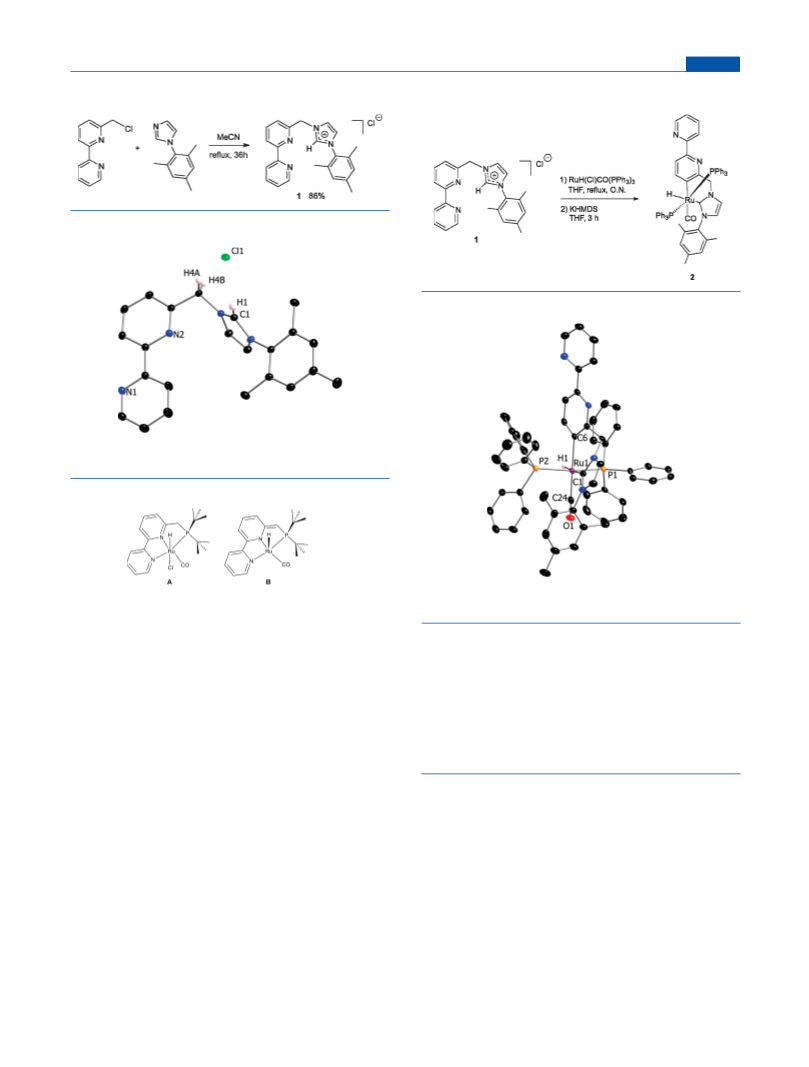
Synthesis of Complex 2. A suspension of 1 (0.127 mmol, 50 mg)
and RuH(Cl)CO(PPh3)3 (0.254 mmol, 242 mg) in dry THF (2.5 mL)
was refluxed under a N2 atmosphere overnight. The reaction mixture
was allowed to cool slowly to room temperature. A yellow precipitate
appeared, and the solvent was decanted. The intermediate product was
washed several times with THF and pentane to yield 102 mg of a
compound in which the NHC part was not coordinated, but all of the
starting ligand was consumed (according to 1H NMR). The above
intermediate product, presumably Ru(PPh3)2(H)(CO) 3-(2,20-bipyr-
idin-6-ylmethyl)-1-mesityl-1H-imidazolium dichloride, was suspended
in dry THF (2.5 mL), and 1 equiv of KHMDS (21 mg) was added,
resulting in a color change to black upon stirring for 3 h at room
temperature. The reaction mixture was filtered, evaporated to dryness,
washed with pentane, and extracted by 4 mL of ether. The resulting
solution was evaporated, and the residue was dissolved in a minimal
amount of benzene. Addition of pentane resulted in precipitation of 2 as
a pure, black solid (29 mg, 22%). Complex 2 was somewhat thermally
unstable. Single crystals suitable for X-ray diffraction were obtained by
layering pentane over a concentrated dichloromethane solution of 2.
31P{1H} NMR (CD2Cl2): 43.22 (d, JP,H = 20 Hz). 1H NMR (300.1
MHz, CDCl3): 8.68 (bd, JH,H = 8 Hz, 1H, o-Py1), 8.57 (bd, JH,H = 8 Hz,
2H, m-Py1), 7.85 (bt, JH,H =7.6 Hz, 1H, m-Py2), 7.79 (bs, 12H,
m-PPh3), 7.72 (bm, 1H, p-Py2), 7.63 (bm, 1H, o-Py2), 7.2ꢀ7.45
(bm, 18H, o,p-PPh3), 7.06ꢀ7.01 (m, 2H, m-Ar + m-Py2), 6.41 (s,
2H, BPyCH2N), 6.37ꢀ6.30 (bm, 2H, imidazole-CHCHN), 2.39 (s, 3H,
p-ArCH3), 2.16 (s, 6H, o-ArCH3) ꢀ12.31(t, JP,H = 20 Hz, 1H, RuꢀH).
IR: ν CdO 1936 cmꢀ1. MS: m/z 1009.35 (M(-H+), calcd m/z 1009.11)
m/z 747.20 (M(-PPh3), calcd m/z 747.83).
Synthesis of Complex 3a. A suspension of 1 (0.077 mmol,
30 mg) and Ru(p-cymene)Cl2(CO) (0.077 mmol, 25.6 mg) in dry
THF (1.5 mL) was refluxed under an argon atmosphere overnight. A red
precipitate appeared, and the solvent was decanted. The intermediate
product was washed several times with THF and pentane and suspended
in dry THF (2.5 mL). One equivalent of KHMDS (0.153 mL of a 0.5 M
solution in toluene) was added to the reaction mixture, and it was
refluxed under an Ar atmosphere for 3 days, resulting in a color change to
black. The reaction mixture was filtered, the solvent was removed under
vacuum, and the residue was washed by pentane and ether and extracted
by THF (1 mL) to give complex 3a as a pure, black-green solid (9 mg,
21%). Single crystals of 3a suitable for X-ray diffraction were obtained by
slow evaporation of a dichloromethane solution. Due to the fact that 3a
was in equilibrium in solution with the cationic 3b, some of the NMR
peaks were broad and overlapping. In addition, the material was
unstable, and therefore so far we were unable to obtain a good 13C
NMR spectrum (at 203.8 K). Single crystals for X-ray diffraction were
obtained from freshly prepared 3.
MS: m/z 576.93 ((M + Na)+, calcd m/z 577.01) m/z 518.98 ((M ꢀ
Clꢀ)+, calcd m/z 519.05). HRMS: m/z 577.0137 (M + Na ꢀ H+, calcd
m/z 577.0112). Note: in all NMR experiments the solution remained
clear and no precipitate was observed. The equilibrium constants are
reproducible and remained the same regardless of whether 3a + 3b was
warmed from low temperature or cooled from ambient temperature.
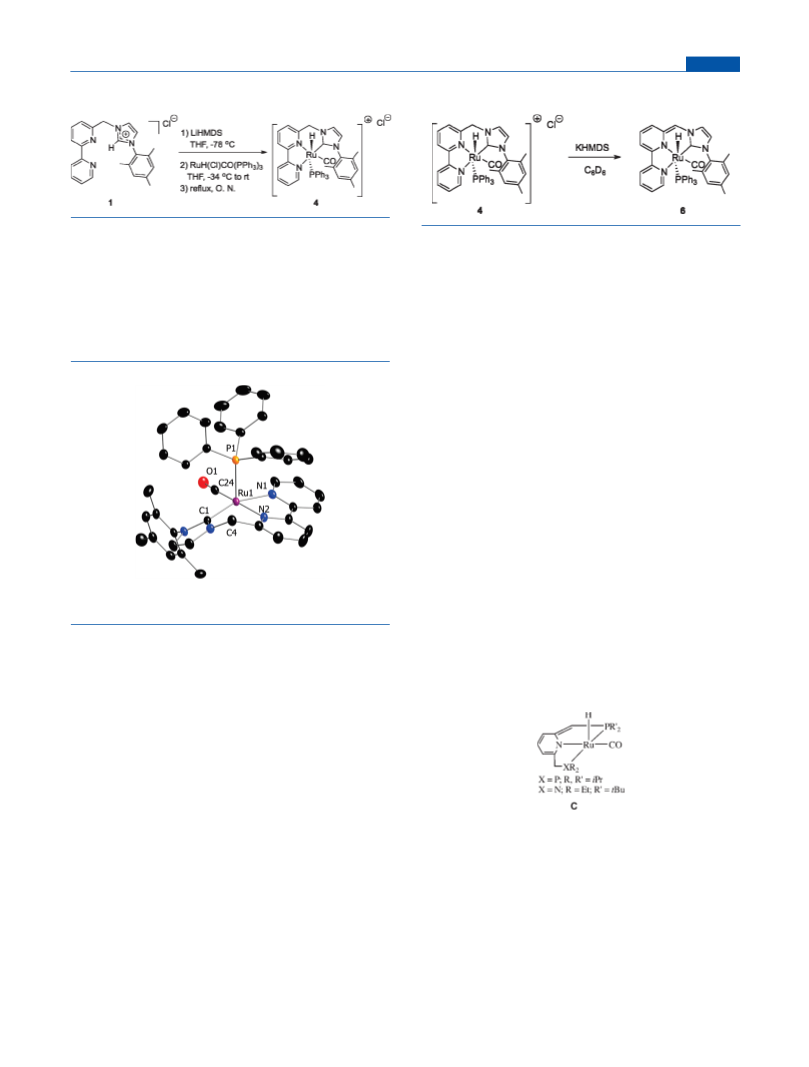
Synthesis of Complex 4. A mixture of solid 1 (500 mg, 1.28
mmol) and LiHMDS (224.5 mg, 1.34 mmol) was cooled to ꢀ78 °C
under Ar; then cold THF (ꢀ78°, 100 mL) was added via cannula, and
the reaction mixture was stirred at the same temperature for 15 min. The
reaction flask was transferred to a calibrated (ꢀ30 οC) ice bath contain-
ing CaCl2/water/dry ice and stirred for 4.5 h. Occasionally the reaction
mixture was allowed to warm to ꢀ28 οC and re-cooled to ꢀ34 οC. Then
the reaction mixture was transferred via a cannula into a flask equipped
with a reflux condenser containing RuHCl(CO)(PPh3)3 (1.219 g, 1.28
mmol) under Ar. The mixture was warmed slowly to room temperature
under Ar and then refluxed under N2 atmosphere overnight. After
cooling to room temperature, the reaction mixture was filtered and
evaporated to dryness, and the resulting solid was washed with pentane
(4 ꢁ 100 mL) and extracted with 20 mL of dichloromethane. The
resulting solution was filtered and concentrated under vacuum until the
appearance of a yellow precipitate. Addition of pentane resulted in
precipitation of an additional, impure yellow precipitate, which was
dissolved in dichloromethane and precipitated again with pentane,
giving 4 as a pure yellow solid (450 mg, 45%).
31P{1H} NMR (CD2Cl2): 24.03(s). 1H NMR (300.1 MHz, CD2Cl2):
8.26 (bm, 2H, o,p-Py1), 8.17 (m, 2H, Ar-H), 8.04 (m, 2H, Ar-H) 7.88
(t, 1H, JH,H = 7.3 Hz, p-Py2), 7.35 (bm, 4H, Ar-H), 7.2 (m, 6H,
m-Ph3P), 7.08 (m, 1H, Ar-H), 6.85ꢀ7 (bm, 9H, o,p-Ph3P), 6.45 (d, 1H,
JH,H = 15.6 Hz, BPyCHHN), 4.45 (d, 1H, JH,H = 15.6 Hz, BPyCHHN),
2.65 (s, 3H, p-ArCH3), 2.05 (s, 3H, o-ArCH3), 1.87 (s, 3H, o-ArCH3),
ꢀ7.96 (d, 1H, JH,P =106 Hz,RuH). 13C{1H} NMR (125.75 MHz,
CD2Cl2): 206.63 (d, JP,C = 6.2 Hz,RuCO), 185.05 (bs, Ru-C NHC),
155.37 (s, imidazole-CHCHN), 154.42 + 154.41 (s, o-Py1+ o-Py2), 154.21
(s,Ar-C-N), 152.27 (s, o-Py1), 136.35 (d, JP,C = 11.3 Hz, PCAr), 133.05
(s,o-Ar),132.81(s,o-Ar),132.54(d,JP,C =45Hz,m-CArP), 126.29 (s, Ar CH),
125.75 (s, Ar CH), 124.88 (s, Ar CH), 122.70 (s, imidazole-CHCHN),
122.17 (s, Ar CH), 121.29 (s, imidazole-CHCHN), 28.94 (s, BPyCH2N),
20.89 (s, p-ArCH3), 18.96 (s, o-ArCH3), 18.70 (s, o-ArCH3). IR: ν CdO
1932 cmꢀ1. HRMS: m/z 747.1838 (M+, calcd m/z 747.1827).
Synthesis of Complex 5 from Complex 4 by Chloride
Exchange with BArFꢀ. To an ethereal suspension (5 mL) of 4
(30 mg, 0.038 mmol) was added 1 equiv of NaBArF (33.7 mg, 0.038 mmol)
under a nitrogen atmosphere. The reaction mixture was stirred for 1 h at
room temperature, and then the formed NaCl was filtered off and the
solvent was removed under vacuum, to yield 53.3 mg (87%) of 5 as a
pure, red solid. The spectral data are essentially identical to that of 4
except for BArF peaks. Single crystals suitable for X-ray diffraction were
obtained by layering pentane over a concentrated ethereal solution of 5.
Synthesis of Complex 6. A 5.7 mg amount of 4 (0.02 mmol) was
suspended in 1 mL of C6D6, and 1 equiv of KHMDS (4.0 mg) was added
to generate a dark green/black complex, 6. Filtration into a J.Young
1H NMR (300.1 MHz, CD2Cl2, 203.8 K) of a solution of 3a + 3b:
Peaks assigned to 3a: 9.10 (d, 2H, JH,H = 5 Hz, o-Py), 8.28 (m, 4H, Ar-
H), 8.21 (m, 3H, Ar-H), 8.12 (m, 3H, Ar-H), 7.91 (m, 2H, Ar-H), 7.71
(d, 2H, JH,H = 2 Hz, Ar-H), 7.31 (m, 3H, Ar-H), 6.99 (m, 2H, Ar-H),
6.97 (m, 2H, Ar-H), 6.93 (m, 1H, Ar-H), 6.76 (m, 1H, Ar-H), 5.72 (bs,
4H, BPyCH2N), 2.34 (m, 12H, ArCH3), 2.2 (m, 9H, ArCH3), 1.51 (m,
6H, ArCH3). Peaks assigned to 3b: 9.34 (d, 1H, JH,H = 5.5 Hz,
o-Py1CH), 7.44 (m, 1H, Ar-H), 7.20 (m, 1H, Ar-H), 7.13 (m, 1H,
Ar-H), 7.09 (m, 1H, Ar-H), 5.80 (bs, 1H, BPyCHHN), 5.34 (bs, 1H,
BPyCHHN). Spectral features of solid 3a: IR: ν CdO 1928.3 cmꢀ1
.
3831
dx.doi.org/10.1021/om200367j |Organometallics 2011, 30, 3826–3833

 Fogler, Eran
Fogler, Eran