Porter and Halasyamani
Table 1. Crystallographic Data for Pb3Te2O6Cl2
Table 2. Atomic Coordinates for Pb3Te2O6Cl2
fw
1043.67
atom
x
y
z
Ueqa (Å2)
space group
a (Å)
b (Å)
C2/m (No. 12)
16.4417(11)
5.6295(4)
10.8894(7)
103.013(10)
982.02(11)
4
Pb(1)
Pb(2)
Pb(3)
Te(1)
Te(2)
Cl(1)
Cl(2)
O(1)
0.26200(6)
0.02126(6)
0.16092(7)
0.10312(10)
0.37188(10)
0.3179(4)
-0.0410(5)
0.1309(8)
0.0000
0.3935(13)
0.2913(9)
0.0000
0.0000
0.5000
0.5000
0.5000
0.5000
0.5000
0.263(3)
0.288(4)
0.5000
0.262(3)
0.20191(9)
0.20025(9)
0.39346(10)
0.05086(15)
0.40736(15)
0.0993(6)
0.3016(8)
0.1830(12)
0.0000
0.0113(4)
0.0108(4)
0.0145(4)
0.0088(4)
0.0088(4)
0.0166(14)
0.0252(17)
0.016(3)
c (Å)
â (deg)
V (Å3)
Z
temp (°C)
λ (Å)
25.0(2)
0.71073
O(2)
O(3)
O(4)
0.024(5)
0.018(2)
0.018(2)
F (g cm-3
µ (cm-1)
R(F)a
)
7.059
0.5810(19)
0.3859(13)
576.65 cm-1
0.0617
Rw(F2)b
0.158
a Ueq is defined as one-third of the trace of the orthogonalized Uij tensor.
a R ) Σ||Fo| - |Fc||/Σ|Fo|. b Rw ) [Σw(|Fo | - |Fc |)2/Σw(Fo )2]1/2
.
2
2
2
Table 3. Summary of Crystallographic Powder X-ray Diffraction and
Refinement Data for Pb3Te2O6Cl2 and Pb3TeO4Cl2
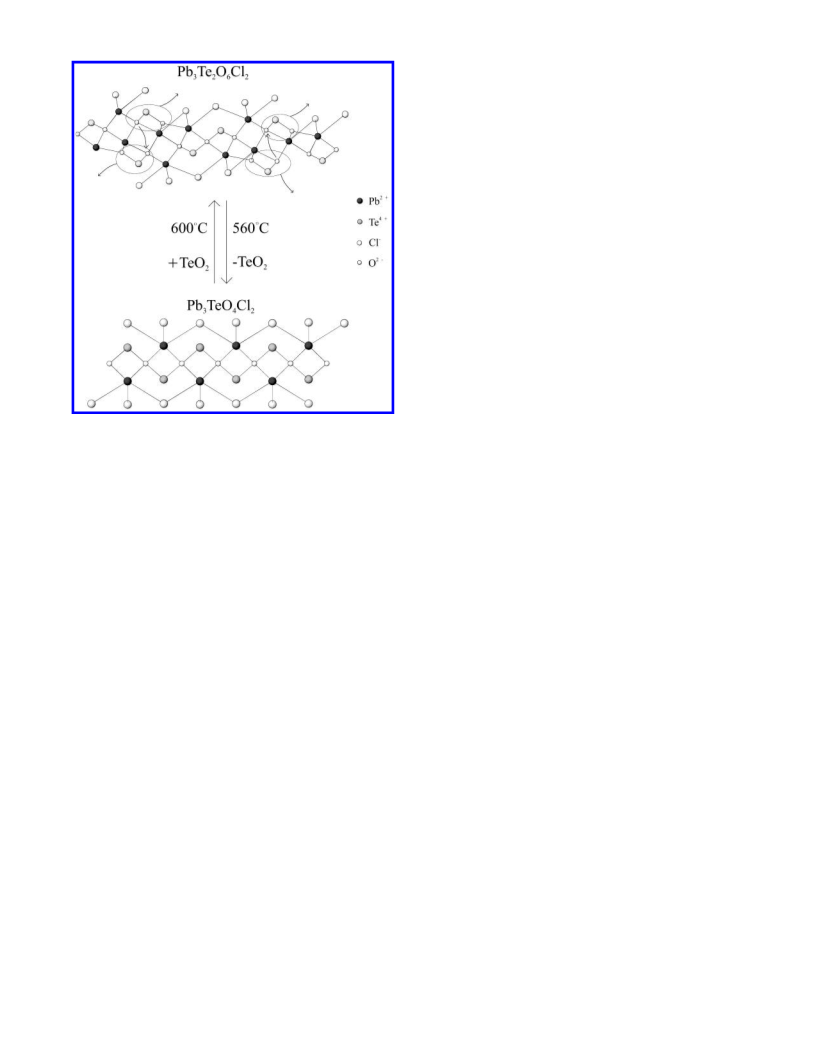
that were subsequently evacuated and sealed. For Pb3Te2O6Br2 (Pb3-
TeO4Cl2 and Pb3TeO4Br2) the tubes were heated to 575 °C (650
°C) for 1 day and cooled at a rate of 6 °C h-1 to room temperature.
With Pb3Te2O6Br2 (Pb3TeO4Cl2 and Pb3TeO4Br2) off-white (yellow)
polycrystalline powders were recovered.
Pb3Te2O6Cl2
Pb3TeO4Cl2
a (Å)
b (Å)
c (Å)
16.4349(5)
5.6255(1)
10.8802(3)
103.050(1)
979.94(1)
C2/m (No. 12)
1380
2.55
0.131
0.167
0.104
5.576(1)
5.559(1)
12.4929(6)
Single-Crystal Structure Determination. The structure of Pb3-
Te2O6Cl2 was determined by standard crystallographic methods.
For Pb3Te2O6Cl2 a colorless column (0.04 mm × 0.08 mm × 0.20
mm) was used for single-crystal measurements. Room-temperature
intensity data were collected on a Siemens SMART diffractometer
equipped with a 1K CCD area detector using graphite- monochro-
mated Mo KR radiation. A hemisphere of data was collected using
a narrow-frame method with scan widths of 0.30° in ω and an
exposure time of 25 s/frame. The first 50 frames were remeasured
at the end of the data collection to monitor instrument and crystal
stability. The maximum correction applied to the intensities was
<1%. The data were integrated using the Siemens SAINT program,5
with the intensities corrected for Lorentz, polarization, air absorp-
tion, and absorption attributable to the variation in the path length
through the detector faceplate. ψ-scans were used for the absorption
correction on the hemisphere of data. The data were solved and
refined using SHELXS-97 and SHELXL-97, respectively.6,7 All
calculations were performed using the WinGX-98 crystallographic
software package.8 Crystallographic data, atomic coordinates, and
thermal parameters for Pb3Te2O6Cl2 are given in Tables 1 and 2.
Powder Diffraction and Crystal Structure Refinement. The
X-ray powder diffraction data were collected on a Scintag XDS2000
diffractometer at room temperature (Cu KR radiation, θ-θ mode,
flat plate geometry) in the 2θ range 3-110° with a step size of
0.02° and a step time of 10 s. The diffraction patterns were analyzed
using the Rietveld9 method with the FULLPROF program.10 An
asymmetry correction was applied to the low-angle reflections. The
scale was refined initially, followed in subsequent iterations by the
zero point error, cell constants, peak shape parameters, atomic
parameters, and overall isotropic temperature factors. For Pb3Te2O6-
Cl2, a powder refinement was undertaken attributable to the
somewhat large errors observed in the single-crystal refinement.
â (deg)
V (Å3)
space group
observns
ø2
385.9(1)
Bmmb (No. 63)
352
3.25
0.169
0.215
0.119
0.087
a
Rp
b
Rwp
c
Rexp
d
RBragg
0.069
b
2
2 1/2
a Rp ) Σ|Io - Ic|/ΣIo. Rwp ) [Σw|Io - Ic| /ΣwIo ]
. .
c Rexp ) Rwp/(ø2)1/2
d RBragg ) Σ|Ik(obs) - Ik(calc)|/ΣIk(obs) , where Io and Ic are the observed and
calculated integrated intensities, Ik is the Bragg intensity, and w is the weight
derived from an error propagation scheme during the process of the least-
squares refinement.
The peaks were indexed on a monoclinic cell, with the refinements
of the unit cell constants performed using a least-squares method.
The structural refinements were carried out in space group C2/m
(No. 12) with a starting model based on the single-crystal data. A
total of 44 parameters, including 13 profile parameters, were used
in the refinement. For Pb3TeO4Cl2 the peaks were indexed on an
orthorhombic cell, with refinement of the unit cell constants
performed by a least-squares method. The structural refinements
were carried out in space group Bmmb (No. 63) with a starting
model based on the structure of orthorhombic PbBiO2Cl. To model
Pb3TeO4Cl2 with the orthorhombic PbBiO2Cl structure, statistical
disorder must occur between the Pb2+ and the Te4+ cations. A
variety of disorder models are possible; however, the model that
gave the best fit to the data and made the most chemical sense is
to statistically disorder Pb(2) and Te(1) (vide infra). In addition,
although the unit cell is metrically tetragonal, refinements using
higher symmetry tetragonal space groups resulted in large errors.
A total of 20 parameters, including 12 profile parameters, were
used in the refinement. The results of the powder refinements,
atomic coordinates, thermal parameters, and bond distances are
given in Tables 3-5. Pb3Te2O6Br2 and Pb3TeO4Br2 are isostructural
with Pb3Te2O6Cl2 and Pb3TeO4Cl2, respectively. Tables 6 and 7
gives the refined unit cell, space group, hkl, dobs, dcalc, Iobs, and Icalc
for Pb3Te2O6Br2 and Pb3TeO4Br2, respectively.
(5) SAINT, version 4.05 ed.; Siemens Analytical X-ray Systems, Inc.:
Madison, WI, 1995.
(6) Sheldrick, G. M. SHELXS-97-A program for automatic solution of
crystal structures; University of Goettingen: Goettingen, Germany,
1997.
(7) Sheldrick, G. M. SHELXL-97-A program for crystal structure refine-
ment; University of: Goettingen: Goettingen, Germany, 1997.
(8) Farrugia, L. J. WinGX: An integrated system of publically available
windows programs for the solution, refinement, and analysis of single-
crystal X-ray diffraction data. J. Appl. Crystallogr. 1998, 32, 837.
(9) Rietveld, H. M. J. Appl. Crystallogr. 1969, 2, 65.
(10) Rodriguez, J. C. FULLPROF Program: RietVeld Pattern Matching
Analysis of Powder Patterns; ILL: Grenoble, France, 1990.
Infrared Spectroscopy. Infrared spectra were recorded on a
Matteson FTIR 5000 spectrometer in the 400-4000 cm-1 range,
with the sample pressed between two KBr pellets.
Thermogravimetric Analysis. Thermogravimetric measure-
ments were carried out on a TGA 2950 thermogravimetric analyzer
(TA Instruments). The samples were contained within platinum
206 Inorganic Chemistry, Vol. 42, No. 1, 2003

 Porter, Yetta
Porter, Yetta