O. Banzragchgarav et al.
PhytochemistryLetters29(2019)79–83
2. Material and methods
Fr.1E (0.96 g) was separated by ODS-120 T column, eluting with
MeOH-H2O, 4:6 and 6:4 to partitions. Partitions were further separated
and purified by HPLCs [Column: ODS-120 T, Develosil C30-UG-5; mo-
bile phase: a gradient of acetonitrile–H2O (1:4 – 3:2, containing 0.2%
TFA)] to find compounds 9 (1.1 mg), 10 (3.1 mg), 12 (14.9 mg), and 13
(3.8 mg).
2.1. General experimental procedures
The UV spectra were recorded with a Shimadzu MPS-2450 (SHIM-
ADZU, Kyoto, Japan). The 1H NMR (400 MHz) and 13C NMR (100 MHz)
spectra were recorded using a Jeol JNM-AL400 FT-NMR spectrometer
(JEOL, Tokyo, Japan), and the chemical shifts were reported as δ values
with TMS as an internal standard at 30 °C (measured in methanol-d4).
Inverse-detected heteronuclear correlations were measured using
The diethyl ether-soluble fraction (3.73 g) was separated using a
silica gel column chromatography, eluting with a gradient of hexane-
acetone (100:0 – 0:100). Further purification by HPLCs with ODS-SM-
50C-M and Capcell Pak C8 columns eluted with acetonitrile–H2O (1:7 –
1
n
HMQC (optimized for JC-H = 145 Hz) and HMBC (optimized for JC-
H = 8 Hz) pulse sequences with a pulsed-field gradient. The HRFABMS
data were obtained using a Jeol JMS700 mass spectrometer (JEOL)
with a glycerol matrix. Preparative HPLC was performed using a Jasco
2089 with UV detection at 210 nm (JASCO, Tokyo, Japan), using the
following columns: Ultra Pack ODS-SM-50C-M (Yamazen, Osaka, Japan
1:3, containing 0.2% TFA) to give compounds 2 (36.8 mg), 3
(191.0 mg), and 5 (42.4 mg).
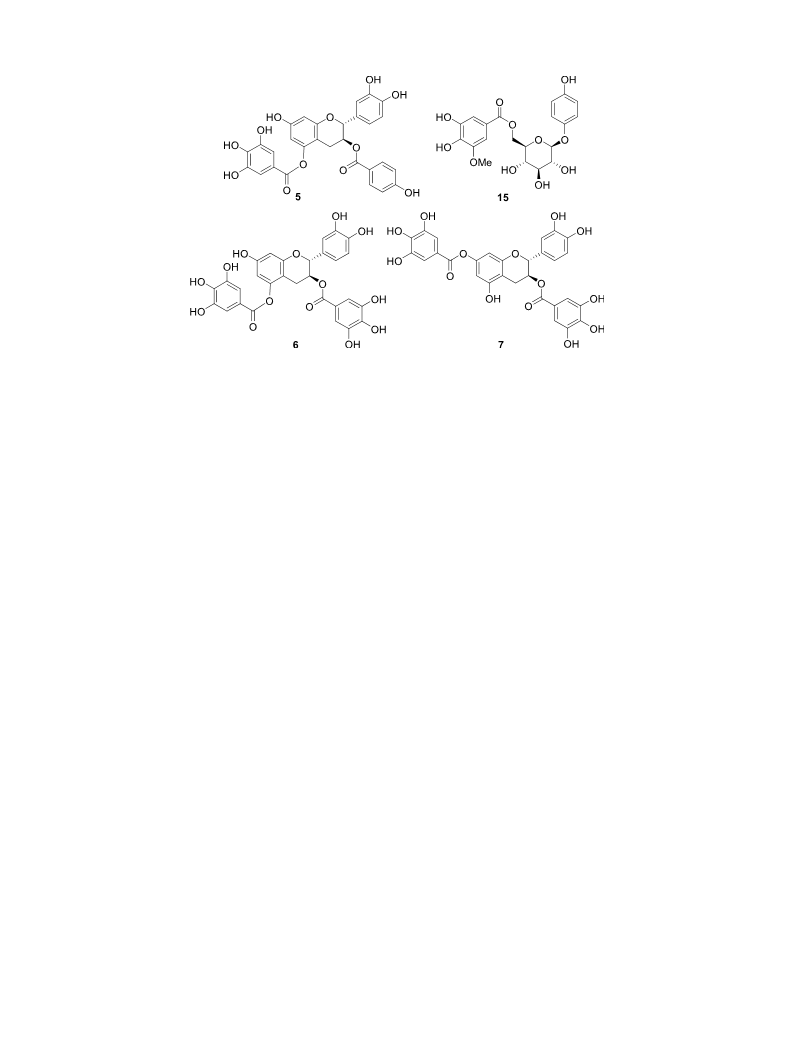
2.3.1. (2R,3S)-3-O-p-hydroxybenzoyl-5-O-galloylcatechin (5)
Colorless amorphous solid; [α]2D5 + 9 (c 2.1, MeOH); UV (MeOH)
max (log ε) 275 (4.39) nm; ECD (c 0.00021, MeOH) ([θ]) 219 (37,000),
λ
37 × 100 mm),
TSKgel
ODS-120 T
(Tosoh,
Tokyo,
Japan,
242 (11,900), 256 (18,000), 285 (–12,600), 401 (2500) nm; HRFABMS
21.5 × 300 mm), and Develosil C30-UG-5 (Nomura Chemical, Aichi,
Japan, 20 × 250 mm), Capcell Pak C8 (Shiseido, Japan, 20 × 250 mm),
and Mightysil RP-18 GP (Kanto Chemicalm Tokyo, Japan,
10 × 250 mm).
(positive-ion mode) m/z 563.1198 [M+H]+ (calcd for C29H23O12
,
563.1189); 1H NMR (methanol-d4, 400 MHz), δ 2.68 (1H, dd, J = 16.5,
6.5 Hz, H-4), 2.83 (1H, dd, J = 16.5, 5.0 Hz, H-4), 5.16 (1H, d,
J = 6.0 Hz, H-2), 5.38 (1H, m, H-3), 6.28 (1H, d, J = 2.5 Hz, H-6), 6.36
(1H, d, J = 2.5 Hz, H-8), 6.75 (2H, overlapping, H-5′ and 6′), 6.76 (2H,
d, J = 8.5 Hz, H-3′′ and 5′′), 6.85 (1H, br s, H-2′), 7.17 (2H, s, H-2′′′ and
6′′′), 7.74 (2H, d, J = 8.5 Hz, H-2′′ and 6′′); 13C NMR (methanol-d4,
100 MHz), δ 24.7 (C-4), 70.6 (C-3), 79.7 (C-2), 101.7 (C-8), 103.7 (C-6),
105.3 (C-4a), 110.6 (C-2′′′ and 6′′′), 114.4 (C-2′), 116.1 (C-3′′ and 5′′),
116.3 (C-5′), 119.2 (C-6′), 120.1 (C-1′′′), 121.9 (C-1′′), 131.0 (C-1′),
132.9 (C-2′′ and 6′′), 140.6 (C-4′′′), 146.4 (C-3′), 146.4 (C-4′), 146.7 (C-
3′′′ and 5′′′), 151.7 (C-5), 156.6 (C-8a), 158.4 (C-7), 163.6 (C-4′′), 166.4
(C-7′′′), 167.2 (C-7′′).
2.2. Plant material
Bergenia crassifolia roots were collected in September 2015 from
Gatsuurt area, Ulaanbaatar, Mongolia, asl; latitude: 48 51′416″ N,
longitude: 107 13′352″ E. A voucher specimen has been deposited in
the herbarium sector of National University of Mongolia. This plant
species was identified by Dr. R. Molor, Mamba Datsan university,
Ulaanbaatar, Mongolia.
2.3. Extraction and isolation
2.3.2. 6′-O-(3′′-O-methylgalloyl) arbutin (15)
Colorless amorphous solid; HRFABMS (positive-ion mode) m/z
439.1247 [M+H]+ (calcd for C20H23O11, 439.1240). 1H NMR (me-
thanol-d4, 400 MHz), δ 3.43 (3H, overlapping, H-Glc-2, Glc-3, and Glc-
4), 3.70 (1H, m, H-Glc-5), 3.85 (3H, s, H-OMe), 4.39 (1H, dd, J = 12.0,
7.0 Hz, H-Glc-6), 4.64 (1H, dd, J = 12.0, 2.5 Hz, H-Glc-6), 4.72 (1H, d,
J = 7.0 Hz, H-Glc-1), 6.58 (2H, d, J = 8.5 Hz, H-3 and 5), 6.92 (2H, d,
J = 8.5 Hz, H-2 and 6), 7.19 (1H, d, J = 2.0 Hz, H-2′), 7.25 (1H, d,
J = 2.0 Hz, H-6′); 13C NMR (methanol-d4, 100 MHz), δ 56.7 (C-OMe),
65.2 (C-Glc-6), 72.0 (C-Glc-4), 75.0 (C-Glc-2), 75.6 (C-Glc-5), 78.0 (C-
Glc-3), 103.8 (C-Glc-1), 106.3 (C-2′), 109.7 (C-6′), 116.7 (C-3 and 5),
119.5 (C-2 and 6), 121.4 (C-1′), 140.8 (C-4′), 146.4 (C-5′), 149.2 (C-3′),
152.4 (C-1), 153.9 (C-4), 168.1 (C-7′).
Air-dried B. crassifolia roots (756 g) were extracted four times with
acetone-H2O (4:1, v/v) at 60°C. Evaporation of the solvent under re-
duced pressure provided a acetone-H2O extract (196 g, 26% from the
roots). The initial separation was performed by means of liquid liquid
partition by resuspending the extract in H2O and the partitioned into
H2O-soluble (195 g), diethyl ether-soluble (3.73 g), and insoluble
(1.28 g) parts. The H2O-soluble fraction was subjected to porous
®
polymer gel (DIAION HP-20, Mitsubishi, Japan) column chromato-
graphy, eluted with H2O-MeOH in a gradient, and obtained Fr.1 A
(H2O, 95 g), Fr.1B (MeOH-H2O, 1:4; 38 g), Fr.1C (MeOH-H2O, 2:3,
35 g), Fr.1D (MeOH-H2O, 3:2, 11 g), Fr.1E (MeOH-H2O, 4:1, 0.96 g),
and Fr.1 F (MeOH, 0.38 g).
A part of Fr.1B (5.0 g) was suspended in MeOH-H2O (1:9) and
centrifuged. Supernatant resulted in 16 subfractions after it was sub-
jected to ODS-SM-50C-M column chromatography, eluting with a gra-
dient of acetonitrile–H2O [1:9 – 2:8, containing 0.2% trifluoroacetic
acid (TFA)]. Then the subfractions were purified by repeated high
performance liquid chromatography [HPLC; Column: ODS-120 T,
Develosil C30-UG-5; and Mightysil RP-18 GP, mobile phase: a gradient
of acetonitrile–H2O (1:19 – 1:3, containing 0.2% TFA)] and isolated
compounds 1 (14.4 mg), 7 (67.2 mg), 8 (1.9 mg), 11 (2.2 mg), 14
(10.9 mg), 16 (44.2 mg), 17 (4.5 mg), 18 (204.9 mg), and 20 (27.8 mg).
A part of Fr.1C (5.0 g) was subjected to ODS-SM-50C-M column
(MeOH- H2O, 1:4) to produce 11 subfractions (Frs. 2 A – 2 K). The
mixture of Frs.2C – 2I was purified by HPLCs [columns: ODS-120 T,
Develosil C30-UG-5, mobile phase: a gradient of acetonitrile–H2O (3:17
– 1:4, containing 0.2% TFA)] to obtain compounds 6 (46.2 mg), 15
(1.3 mg), 21 (54.8 mg), and 22 (18.3 mg).
2.4. Sugar identification
The previous procedure using HPLC for glycosyl moiety identifica-
mixture was concentrated after going through an HP-20 column
(5 × 20 mm) with water (1 mL). Then, each residue was stirred with L-
cysteine methyl ester (5 mg) in pyridine (0.5 mL) (60℃, 1 h), and then
o-tolyl isothiocyanate (10 μL) was added (60℃, 1 h). The reaction
mixtures were analyzed by HPLC (column, Capcelpak C18 UG120, Si-
seido, Tokyo, Japan, 4.6 × 250 mm; mobile phase, acetonitrile-0.2%
TFA in H2O (25:75), 1.0 mL/min; detector, UV at 250 nm), and the
peaks of reactants from 15 (tR 18.3 min), D-glucose (tR 18.3 min), and
L-glucose (tR 16.7 min) were detected.
2.5. In vitro cultivation of the parasites, B. bovis and B. bigemina
Fr.1D (11.0 g) was subjected to ODS-120 T column, eluting with a
gradient of MeOH-H2O (1:3 – 6:4), and then purified by Develosil C30
-
2.5.1. Texas strain of B. bovis and the Argentine strain of B. bigemina
UG-5 column chromatography, eluting with gradient of
acetonitrile–H2O (1:7 – 1:3, containing 0.2% TFA) to yield compounds
4 (17.5 mg) and 19 (11.3 mg).
80

 Banzragchgarav, Orkhon
Banzragchgarav, Orkhon