Table 2 Pd-catalyzed synthesis of arylacetic acid derivatives
bromoacetic esters or a-bromoacetic amides represents a mild
and general approach to arylacetic acid derivatives. The simple
reaction protocol which involves only air-stable chemicals and
does not require absolutely dry solvents makes this reaction a
valuable alternative to the existing protocols, especially for
applications in combinatorial chemistry and drug discovery.
I thank M. Rössig and A. Söthe for technical assistance and
Professor Dr M. T. Reetz for generous support and constant
encouragement.
Method A
Method B
Yield (%)a
Comp. Ar
X
Yield (%)a
3a
3b
3c
3d
3e
3f
3g
3h
3i
3k
3l
3m
6
Phenyl
o-Tolyl
OEt
OEt
OEt
OEt
OEt
OEt
OEt
OEt
OEt
OEt
OEt
OEt
N(C5H10
85 (90)
90 (90)
80 (84)
84 (85)
79 (80c
90 (93)
70 (75)b
67 (74)c
40 (40)b
63 (70)
33 (33)b
31 (42)c
81 (89)
87 (91)
75 (79)
68 (70)
76 (79)
60 (65)
1-Naphthyl
p-MeO-Phenyl
p-Acetylphenyl
p-Tolyl
m-Chlorophenyl
p-Formylphenyl
m-Nitrophenyl
m-AcNH-Phenyl
2-Thienyl
Notes and references
‡ Synthesis of 4-bromobutyl phenylacetate (7): A 100 mL flask was charged
with palladium acetate (67.3 mg, 0.30 mmol), tri-1-naphthylphosphine (371
mg, 0.90 mmol), 4-bromobutyl bromoacetate (2b) (2.74 g, 10.0 mmol) and
an excess of potassium phosphate (10.61 g, 50.0 mmol). The reaction vessel
was purged with argon, a solution of benzeneboronic acid (1.46 g, 12.0
mmol) in THF (40 ml) was added and the reaction mixture was stirred at 20
°C overnight. The reaction slurry was then poured into water (300 mL) and
extracted 33 with 100 mL portions of dichloromethane. The combined
organic layers were dried over MgSO4, filtered, and the volatiles were
removed in vacuo. The residue was purified by fractional distillation. A
colorless oil (2.41 g, 89%) boiling at 91 °C/0.01 mbar was collected and
identified as the desired product. 1H NMR (300 MHz, CDCl3, 25 °C, TMS):
d = 7.33–7.26 (m, 5H), 4.12 (t, 3J (H,H) = 6 Hz, 2H), 3.62 (s, 2H), 3.38
(t, 3J (H,H) = 6 Hz, 2H), 1.87 (m, 2H), 1.79 (m, 2H) ppm; 13C NMR (75
MHz, CDCl3, 25°C, TMS): d = 171.5, 134.0, 129.2, 128.6, 127.1, 63.8,
41.4, 32.9, 29.2, 27.2 ppm; MS (70 eV): m/z (%): 270(6) [M+], 191(4),
179(4), 136(23), 91(100); HRMS: calcd. for C12H15BrO2 [M+]: 270.02555;
found: 270.02546; anal. calcd. for C12H15BrO2 (271.16): C, 53.16; H, 5.58;
N, 0.0; found: C, 52.96; H, 5.65; N, 0.0. The reactions in Table 1 and Table
2 were performed at least twice on 1 mmol scale using 0.05 mL tetradecane
as an internal GC standard. The products were isolated by column
chromatography (SiO2, hexane–ethyl acetate 10+1) and characterized by
means of 1H and 13C NMR as well as by GC-MS.
2-Fluorophenyl
Phenyl
)
7
Phenyl
O(C4H8)Br 72 (90)
68 (72)
Conditions: (A) 1.2 equiv. arylboronic acid, 3 mol% Pd(OAc)2, 9 mol%
P(Nap)3, 5 equiv. K3PO4, 2 equiv. H2O, 20 °C, THF; (B) 1.2 equiv. pinacol
boronate, 3 mol% Pd(OAc)2, 9 mol% P(Nap)3, 5 equiv. K3PO4, 2 equiv.
H2O, 20 °C, THF. a Isolated yields (GC-determined yields in parentheses).
b KF instead of K3PO4. c K2CO3 instead of K3PO4
water had no adverse effect on the reaction outcome so that
special drying of the solvent and the reagents is not required.
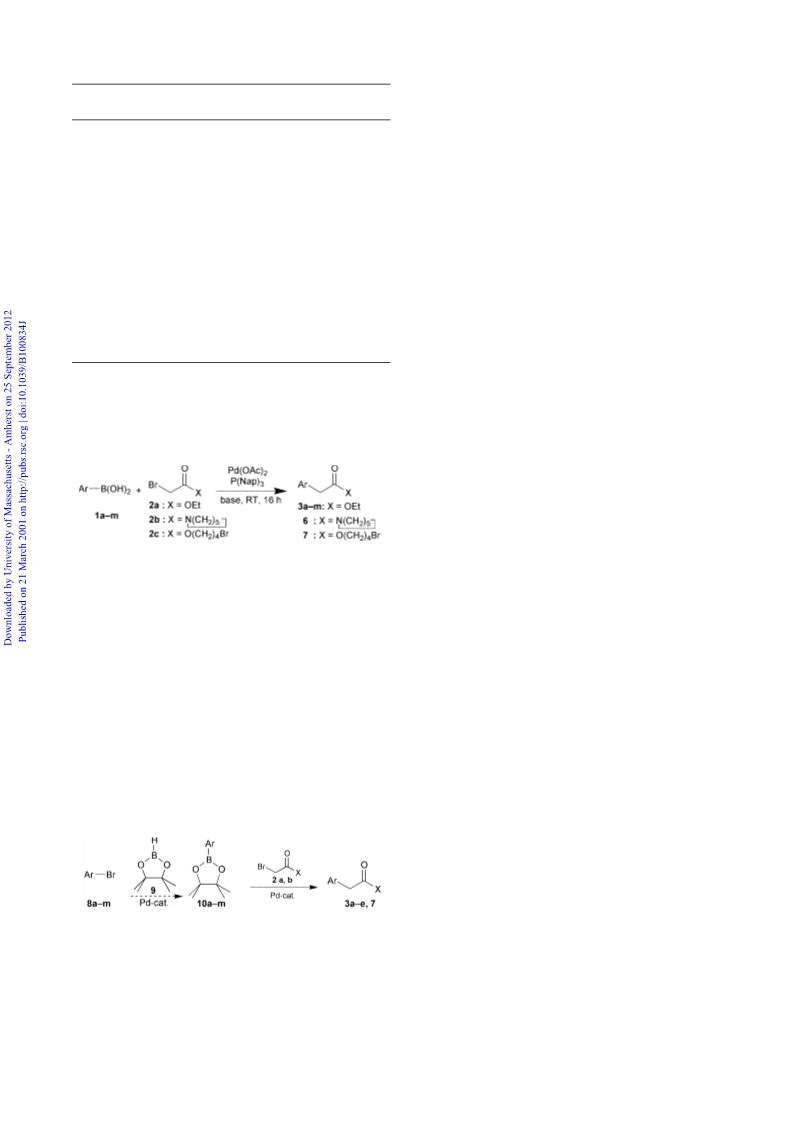
The generality and selectivity of the reaction were investi-
gated using a number of arylboronic acids 1a–m in combination
with several alkyl halides (Scheme 2).
§ The IUPAC name for pinacol boronates and pinacol borane is 2,3-bor-
anediyldioxy-2,3-dimethylbutane.
Scheme 2 Pd-catalyzed synthesis of arylacetic acid derivatives.
1 T. Y. Shen, Angew. Chem., 1972, 84, 512; Angew. Chem., Int. Ed. Engl.,
1972, 11, 460.
As can be seen in Table 2 (Method A), most substrates give
good yields. Electron-poor and electron-rich compounds are
equally suitable for the transformation. Even sterically hindered
compounds (o-tolylboronic acid) or substrates that are some-
times problematic in palladium-catalyzed reactions (nitro- or
heteroarenes) were successfully employed. Moreover, products
containing enolizable keto-groups are readily formed without
any signs of side products arising from undesired aldol
condensations.
Besides ethyl bromoacetate (2a), other bromoacetates (2c) or
-amides (2b) can be used. The high selectivity of the
transformation is demonstrated by the formation of compound
7‡ (Table 2): the arylation of 4-bromobutyl bromoacetate (2c)
takes place exclusively a to the carbonyl group even though a
primary alkyl bromide functionality is present.
2 For common methods such as the hydrolysis of benzylnitriles, the
carbonylation of benzyl halides, or the Willgerodt reaction of acet-
ophenones see: J. March, Advanced Organic Chemistry, Wiley, New
York, 4th Edn., 1992, pp. 1281–1282; further methods: (a) T. Zincke,
Chem. Ber., 1869, 2, 738; (b) R. Quelet and J. Gavarret, Bull. Soc. Chim.
Fr., 1950, 1075; (c) J. B. Woell and H. Alper, Tetrahedron Lett., 1984,
25, 3791; (d) Prileshajew, Zh. Russ. Fiz.-Chim. O-va, 1910, 42, 1395.
3 (a) W. W. Leake and R. Levine, J. Am. Chem. Soc., 1959, 81, 1627; (b)
J. F. Fauvarque and A. Jutand, J. Organomet. Chem., 1979, 177, 273; (c)
F. Orsini and F. Pelizzoni, Synth. Comm., 1987, 17, 1389.
4 M. Kosugi, Y. Negishi, M. Kameyama and T. Migita, Bull. Chem. Soc.
Jpn., 1985, 58, 3383.
5 K. Okuro, M. Furuune, M. Miura and M. Nomura, J. Org. Chem., 1993,
58, 7606.
6 (a) M. van Leeuwen and A. McKillop, J. Chem. Soc., Perkin Trans. 1,
1993, 2433; (b) M. F. Semmelhack, B. P. Chong, R. D. Stauffer, T. D.
Rogerson, A. Chong and L. D. Jones, J. Am. Chem. Soc., 1975, 97, 2507;
(c) S. G. Lias and P. Ausloos, J. Am. Chem. Soc., 1977, 99, 4833; (d) M.
Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121, 1473.
7 (a) C. Carfagna, A. Musco and C. Sallese, J. Org. Chem., 1991, 56, 261;
(b) T. Sakamoto, Y. Kondo, K. Masumoto and H. Yamanaka,
Heterocycles, 1993, 36, 2509; (c) F. Agnelli and G. A. Sulikowski,
Tetrahedron Lett., 1998, 39, 8807.
Many functionalized pinacol boronates§ (10) are conven-
iently accessible from aryl halides (8) and bispinacol diboron9
or pinacol borane (9).10 We thus considered it to be important to
extend our reaction to this substrate class (Scheme 3).
8 (a) J. Chaussard, J.-C. Folest, J.-Y. Nédélec, J. Périchon, S. Sibille and
M. Troupel, Synthesis, 1990, 369; (b) M. Durandetti, J.-Y. Nédélec and
J. Périchon, J. Org. Chem., 1996, 61, 1748; (c) J.-C. Folest, J. Périchon,
J. F. Fauvarque and A. Jutand, J. Organomet. Chem., 1988, 342, 259.
9 (a) T. Ishiyama, M. Murata and N. Miyaura, J. Org. Chem., 1995, 60,
7508; (b) T. Ishiyama, Y. Itoh, T. Kitano and N. Miyaura, Tetrahedron
Lett., 1997, 38, 3447.
Scheme 3
10 (a) M. Murata, T. Oyama, S. Watanabe and Y. Masuda, J. Org. Chem.,
1997, 62, 6458; (b) M. Murata, T. Oyama, S. Watanabe and Y. Masuda,
J .Org. Chem, 2000, 65, 164.
11 M. Sato, N. Miyaura and A. Suzuki, Chem. Lett., 1989, 1405.
12 N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457.
13 (a) S. Yamaguchi, S. Ohno and K. Tamao, Synlett, 1997, 10, 1199; (b)
M. Moreno-Manas, M. Perez and R. Pleixats, J. Org. Chem., 1996, 61,
2346.
We were pleased to find that the best conditions for the
conversion of the boronic acids turned out also to be optimum
conditions for pinacol boronate and that the reaction usually
gives similar yields for both starting materials (Table 2, ‘method
B’).
In summary, the disclosed palladium-catalyzed cross-cou-
pling reaction between arylboronic acids or esters and a-
670
Chem. Commun., 2001, 669–670

 Goossen
Goossen